
Wie kann eine Phänotypisierung bei der Diabetesbehandlung helfen? Professor Andreas Pfützner berichtet über seine Erfahrungen in der Praxis. Lesen Sie hier und in einer der nächsten Ausgaben, wie eine personalisierte und individuelle Therapie aussehen könnte.
Typ-2-Diabetes ist eine hochkomplexe multifaktorielle Erkrankung, die aus mehreren ineinandergreifenden Grundstörungen (ß-Zelldysfunktion, Insulinresistenz, hormonelle Hyperaktivität des viszeralen Fettgewebes und chronisch systemischer Inflammation) besteht. Die seit Jahrhunderten praktizierte Diagnosestellung ausschließlich über die Urin- und Blutzuckererhöhung, die letztlich nur ein Symptom der Krankheit darstellt, erfasst das Krankheitsbild nur sehr oberflächlich [King 2003, ElSayed 2023]. Des Weiteren führt die aktuelle leitliniengerechte Therapie mit praktisch ausschließlicher Fokussierung auf die Normalisierung des Blutzuckers und seines Surrogat-Parameters HbA1c [ElSayed 2023] dazu, dass Diabetes mellitus als chronisch progrediente Erkrankung angesehen wird, bei der selbst bei Erreichung der Therapieziele ein relativ früher Tod durch makrovaskuläre oder mikrovaskuläre Ereignisse praktisch nicht zu vermeiden ist [ACCORD 2008, Duckworth 2009, ElSayed 2023, Patel 2007]
Auf der Basis einer umfangreichen Studienerfahrung wurde in meiner Praxis eine Methode zur Phänotypisierung und konsekutiven personalisierten Diabetestherapie entwickelt, die ich an dieser Stelle gerne vorstellen und zur Diskussion stellen möchte. Wichtig vorab – dieses Diskussionspapier hat keinen missionarischen Hintergrund. Ich möchte unseren individualisierten Ansatz zur Diabetestherapie, der auf einer mehr als 30-jährigen klinischen und Studien-Erfahrung basiert und den ich seit mehr als 15 Jahren erfolgreich in der Praxis anwende, einem breiteren Kollegenkreis vorstellen und dazu einladen, es gegebenenfalls und bei Interesse einfach mal auszuprobieren bzw. gerne auch zu kommentieren.
In diesem Teil 1 wird der Hintergrund und die Vorgehensweise bei der Phänotypisierung beschrieben. Im nachfolgenden Teil 2 wird die Umsetzung der Phänotypisierung in eine individuelle Diabetestherapie vorgestellt und gezeigt, welche Ergebnisse wir in der Praxis mit diesem Konzept bisher erzielen konnten.
Physiologische Grundlagen
Die biologischen Vorgänge im Organismus, die heute eine wichtige Grundlage der Pathophysiologie des Typ-2-Diabetes sind, waren vor vielen Jahrtausenden durchaus ein Überlebensvorteil. Es kann davon ausgegangen werden, dass die Menschheit in der Steinzeit, als unsere Vorfahren noch in Höhlen lebten und urzeitliche Tiere jagten, nicht immer regelmäßig und ausreichend Nahrung zur Verfügung hatte. Dadurch hatten Individuen einen Überlebensvorteil – und haben sich genetisch durchgesetzt – die in der Lage waren, zu Zeiten eines Nahrungsmittelüberflusses so viel Energie wie möglich in Form von Fettgewebe abzuspeichern, um dann in Zeiten des Hungerns davon zehren zu können. Die Bildung von Fettgewebe im Körper ist physiologischer Weise eine ausschließliche Domäne der Insulinwirkung [Hauner 1987]. Insulin stimuliert bekanntlich die Ausdifferenzierung mesenchymaler Stammzellen zu Präadipozyten (siehe Abb. 1) [Rosen 2006 und 2014]. Diese sind wiederum die Quelle zahlreicher sogenannter "Adipokine", die trotz hoher Diversität in ihren Wirkungen eine biochemische Eigenschaft gemeinsam haben: sie wirken gegen Insulin [Das 2023, Rosen 2006 und 2014]. Hierdurch entsteht eine Art metabolischer Insulinresistenz und der Insulinbedarf zur Kontrolle des Blutzuckers steigt an. Die Produktionskapazität der pankreatischen ß-Zellen ist sehr hoch und konsequenterweise wird nun als Antwort auf den erhöhten Insulinbedarf mehr Insulin produziert, dass dann seinerseits zur weiteren Ausdifferenzierung mesenchymaler Stammzellen führen kann. Schlussendlich erlauben diese physiologischen Zusammenhänge dem Körper mehr Insulin zu tolerieren und mit seiner Hilfe Fettgewebe als Energiespeicher zu generieren, ohne gleichzeitig negative Auswirkungen auf den Blutzuckerspiegel zu erfahren. Ein ohnmächtiger Mensch in der Hypoglykämie kann schließlich auch keine Nahrung zu sich nehmen. Tatsächlich leben wir zumindest in der westlichen Welt aktuell in dauerhaften Zeiten des Nahrungsmittelüberflusses. In der Konsequenz der vorhandenen evolutionären Vorgaben erlebt die zivilisierte Welt seit mehreren Jahrzehnten eine Adipositas-Welle, die es vorher in der Historie der Menschheit nie gegeben hat [World Obesity Atlas 2022].
Leider haben die Adipokine unter anderem negative Auswirkungen auf den Blutdruck. Ein prominenter Vertreter ist hier Angiotensin II, das unkontrolliert von den Präadipozyten gebildet und ausgeschüttet wird und den Blutdruck nach oben treibt [Hall 2019]. Um Trigylzeride einlagern zu können, induzieren einige Adipokine die Bildung von freien Fettsäuren und Trigylyzeriden, und es kommt in dieser Situation daher häufig zu Hypertriglyzeridämie und niedrigen HDL-Spiegeln [Lafontan 2008]. Das durch Bluthochdruck und Dyslipidämie bereits erhöhte kardiovaskuläre Risiko (siehe Abb. 1), führt schon unabhängig vom Diabetes dazu, dass die damit assoziierten fatalen Erkrankungen (Herzinfarkt, Schlaganfall etc.) zu den Haupttodesursachen in der westlichen Welt gezählt werden müssen [WHO 2020]. Der ebenfalls Insulinresistenz-bedingte Ausfall der bekannten vasoprotektiven Insulinwirkung (anti-oxidative NO-Sekretion) [Forst 2009] trägt das seine zusätzlich dazu bei.
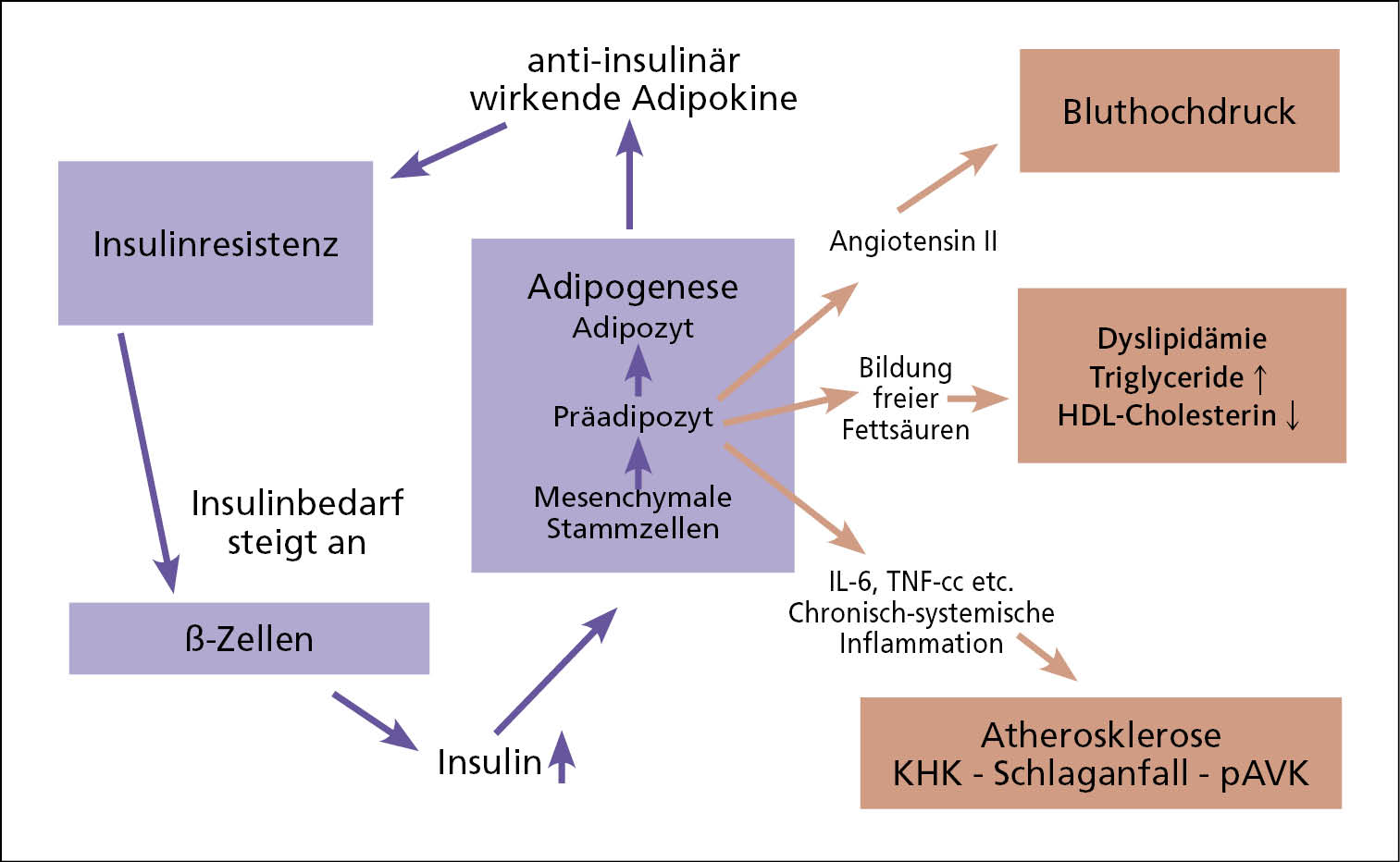 Abb. 1: Physiologische Vorgänge bei der Gewichtszunahme und damit assoziierte mögliche pathologische Folgen.
Abb. 1: Physiologische Vorgänge bei der Gewichtszunahme und damit assoziierte mögliche pathologische Folgen.
Ein weiterer, das makrovaskuläre Risiko erhöhender Faktor ist die Entwicklung einer chronischen Aktivierung des Immunsystems (chronisch-systemische Inflammation) auf Grundlage der Stammzellausdifferenzierung im Fettgewebe [Andersson 2008, Murdolo 2006]. Immer wenn im Körper Ausdifferenzierungsprozesse stattfinden, kommt es in sehr geringem Umfang auch zur unvollständigen Ausdifferenzierung von Stammzellen mit einem gewissen Entartungsrisiko, und damit zur gelegentlichen Bildung mutagener Zellen. Damit diese keinen Schaden anrichten können ist das Immunsystem alarmiert und aktivierte Makrophagen wandern ins Fettgewebe ein, die mutierte Zellen erkennen und vernichten können [Dandona 2004a und 2004b]. Da das Immunsystem aber nicht ausschließlich lokal aktiviert werden kann, sind in dieser Situation alle Monozyten/Makrophagen im Körper aktiviert und damit auch die Immunzellen, die im Kreislauf zirkulieren [Ghanim 2004, Ringeis 2015]. Diese haben gegebenenfalls jedoch bereits oxidiertes LDL-Cholesterin opsonisiert [Mushenkova 2021, Orekov 2020]. Die durch weitere Triggermechanismen (z.B. Hochdruck, Hyperglykämie etc.) induzierte Penetration dieser aktivierten lipid-beladenen Monozyten/Makrophagen in die Gefäßwand ist die immunologische Basis für die Entstehung einer Atherosklerose [Boyle 2003].
Die geschilderten Zusammenhänge lassen bereits erahnen, warum Menschen mit Übergewicht und vor allem während einer Gewichtszunahme zu Hochdruck, Fettstoffwechselstörungen, Insulinresistenz und Atherosklerose neigen [Freeman 2023]. Viel ernster wird die Situation allerdings, wenn nun auch noch ein Typ-2-Diabetes mellitus dazukommt.
Pathophysiologische Aspekte des Typ-2-Diabetes mellitus
Typ-2-Diabetes mellitus beruht nach allen verfügbaren aktuellen genetischen Untersuchungen hauptsächlich auf erblichen Störungen der ß-Zelldysfunktion [Alford 1998, O’Rahilly 1986]. Während in der Allgemeinheit häufig die Botschaft verbreitet wird, dass eine ungesunde Lebensführung zu Diabetes führt, muss festgestellt werden, dass nur wer die Diabetes-Gene in sich trägt, tatsächlich auch einen Typ-2-Diabetes entwickeln wird. Ansonsten wird man zwar adipös und bekommt wahrscheinlich neben Herz-Kreislauf-Symptomen auch irgendwann noch orthopädische Probleme. Durch eine gesunde Lebensführung lässt sich allerdings auch nach meiner Erfahrung die Manifestation des Diabetes zeitmäßig deutlich nach hinten verschieben. Beim Typ-2-Diabetes stehen mehrere Sekretionsstörungen der ß-Zelle im Vordergrund: zunächst fallen nach aktueller Datenlage die physiologische Pulsatilität der Insulinsekretion sowie die erste Insulinantwort auf die Mahlzeit aus (zeitliche Sekretionsstörung) [Brunzell 1976, Lang 1981, Polonsky 1988]. Anstatt sechsmal pro Stunde einen Insulinpeak auszuschütten, erfolgt die Insulinsekretion in einem "Steady-Flow" (Stadium I der ß-Zelldysfunktion" [Wahren 2012, Pfützner 2004a]). Hierdurch fällt die protektive Wirkung von Insulin in der Mikrozirkulation aus [Forst 2009]. Dies könnte ein Grund dafür sein, warum auch blutzuckermäßig gut eingestellte Menschen mit Diabetes nach ausreichender Diabetesdauer Mikrozirkulationsstörungen entwickeln können und dies insbesondere, wenn zusätzliche andere Risikofaktoren vorhanden sind [Toyama 2023]). Aufgrund des Insulinresistenz-vermittelten Insulin-Mehrbedarfs kommt es bekanntermaßen zur "Hyperinsulinämie" (quantitative Sekretionsstörung, Stadium II der ß-Zelldysfunktion) [Pfützner 2004a], was in der Folge zur Erschöpfung der Spaltungskapazitäten in der ß-Zelle führt, und konsekutiv eine Ausschüttung von Proinsulin neben Insulin zur Folge hat (qualitative Sekretionsstörung; Stadium III der ß-Zelldysfunktion) (Abb. 2) [Pfützner 2004a].
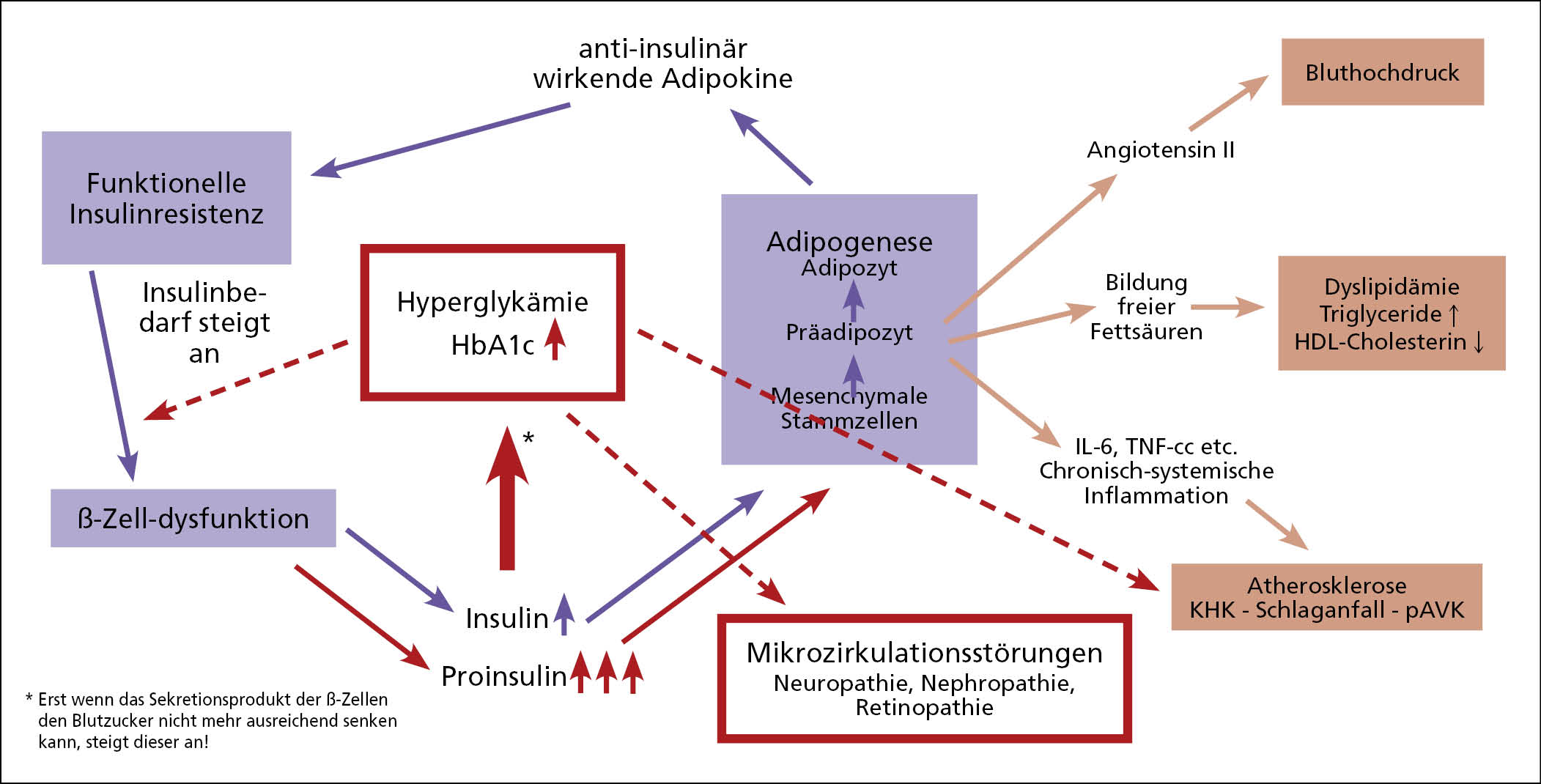 Abb. 2: Pathophysiologische Vorgänge beim Typ-2-Diabetes.
Abb. 2: Pathophysiologische Vorgänge beim Typ-2-Diabetes.
Proinsulin ist ein nicht-physiologisches Hormon, dass ebenfalls den Blutzucker senken kann, allerdings nur mit 10 - 20 % der Effektivität von Insulin [Galloway 1992]. Gleichzeitig hat es die gleiche lipogenetische Funktionalität wie Insulin [Pfützner 2017], wodurch die negativen Wirkungen der Adipokine auf den Körper weiter verstärkt werden können. Die blutzuckersenkende Wirkung von Proinsulin kann in dieser Situation dazu beitragen eine Normoglykämie aufrecht zu erhalten, während die übrigen pathologischen Vorgänge unvermindert voranschreiten. Ich habe diesen widersprüchlichen Zustand eines "normoglykämischen" Diabetes schon vor fast 2 Jahrzehnten gemeinsam mit anderen Autoren beschrieben, und damals als "Kardiodiabetes" oder "Gefäßdiabetes" bezeichnet [Kintscher 2006]. In dieser Phase findet im Körper gewissermaßen ein Wettlauf statt: Reicht die Zeit noch für die Blutzucker-basierte Diagnose eines Diabetes mellitus aus, oder kommt es vorher bereits zu einem fatalen makrovaskulären Ereignis. Die beschriebenen Zusammenhänge erklären auch, warum bei Erstdiagnose eines Typ-2-Diabetes mellitus bei einer Vielzahl der betroffenen Menschen bereits manifeste atherosklerotische Gefäßveränderungen diagnostiziert werden [Ruigomez 1998, ElSayed 2023].
Eine Hyperglykämie erhöht ihrerseits den Insulinbedarf und kurbelt den Insulinbedarf weiter an. Hierdurch wird der Circulus vitiosus dieser Pathophysiologie noch einmal beschleunigt, was die Notwendigkeit einer guten Blutzuckereinstellung unterstreicht. Wird der Blutzucker nicht konsequent normalisiert, führt die Glukosetoxizität zu einer deutlichen Beschleunigung bei der Entwicklung der bekannten mikrovaskulären und makrovaskulären Spätschäden.
Warum Phänotypisierung?
Die beschriebenen Grundstörungen (ß-Zelldysfunktion, Insulinresistenz, Aktivität des viszeralen Fettgewebes, chronisch systemische Inflammation (CSI)) können in unterschiedlichen Ausprägungen und Schweregraden vorliegen, die durchaus zu unterschiedlichen klinischen Phänotypen führen (Abb. 3).
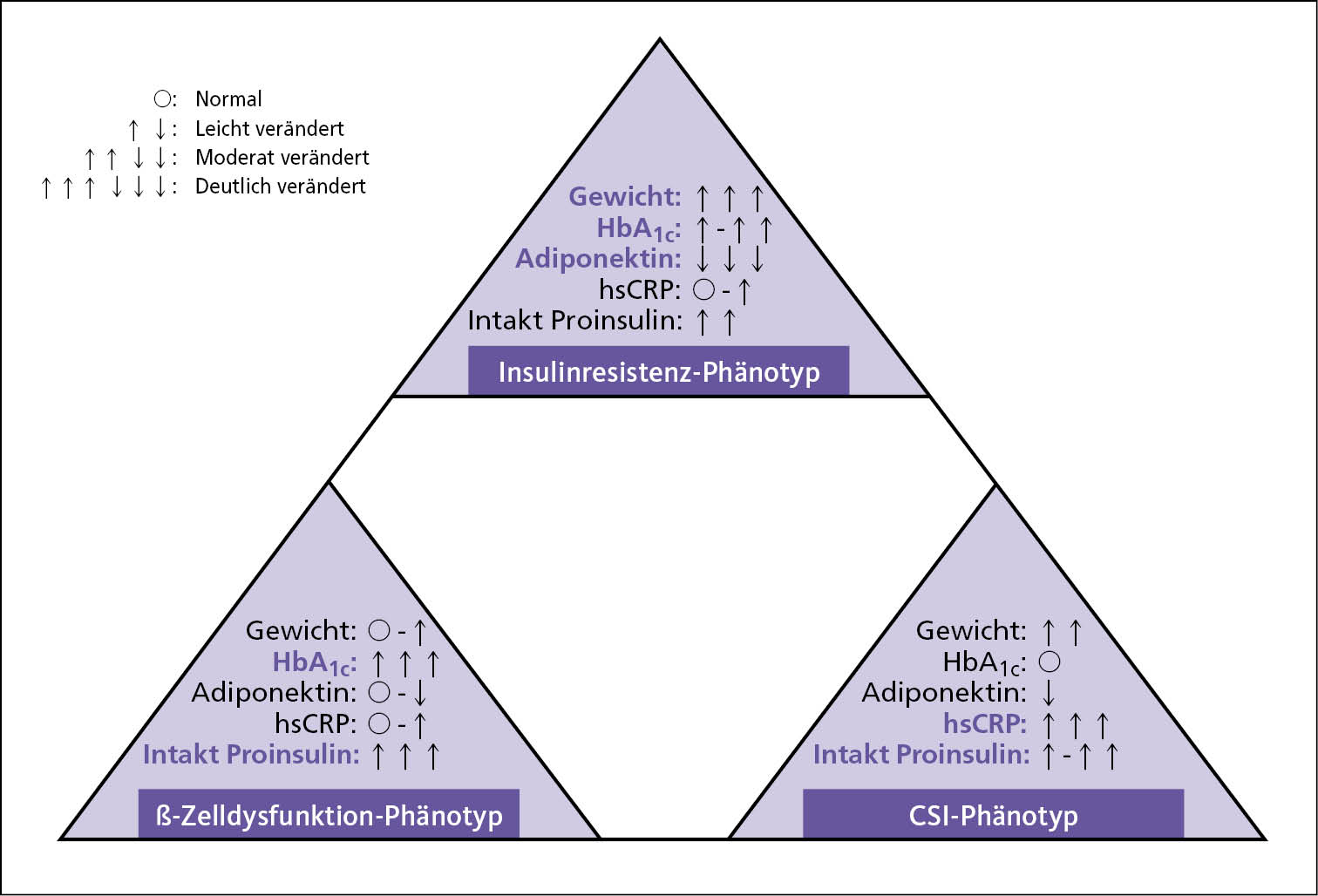 Abb. 3: Funktionelle Extrem-Phänotypen beim Typ-2-Diabetes (mit Hervorhebung der Leitindikatoren).
Abb. 3: Funktionelle Extrem-Phänotypen beim Typ-2-Diabetes (mit Hervorhebung der Leitindikatoren).
So gibt es als einen Extremfall den normoglykämischen Menschen mit "Kardiodiabetes", der häufig leicht übergewichtig ist und sich klinisch mit arteriellem Hochdruck, Fettstoffwechselstörung und oft auch Hyperurikämie präsentiert (CSI-getriebener Phänotyp). Dieser Phänotyp wird aktuell nicht erkannt und ausschließlich symptomatisch behandelt.
Der vor allem durch die ß-Zelldysfunktion getriebene Phänotyp zeigt eine schlechte Ansprache auf die leitliniengerechte Diabetes-Therapie. Die betroffenen Menschen sind häufig eher schlank oder nur leicht übergewichtig und haben meist so lange schlechte Blutzucker- und HbA1c-Werte bis bei der leitliniengerechten Therapieeskalation endlich Insulin eingesetzt wird.
Der Insulinresistenz-getriebene Phänotyp präsentiert sich mit deutlichem Übergewicht, schwer behandelbarem Hochdruck und oft auch schon mit manifester und systemischer Atherosklerose.
Der Phänotyp eines jeden Menschen mit Diabetes liegt nach meiner Erfahrung ganz individuell zwischen diesen drei Extremen und es erscheint schwer nachvollziehbar, dass man mit einer glukozentrischen und HbA1c-fixierten eskalierenden Standardvorgehensweise (Diät & Lebensstil -> Metformin ->Metformin & ein weiteres Antidiabetikum -> Metformin & zwei weitere Antidiabetika -> Insulin & weitere Antidiabetika, [BÄK 2021]) dieses Krankheitsbild so effizient behandeln kann, dass mikrozirkulatorische Spätschäden und vor allem auch makrovaskuläre Ereignisse verhindert werden können. In jedem Fall führt die leitliniengerechte HbA1c-gesteuerte Standardtherapie nach aktueller Datenlage nicht wirklich zur Reduktion der Haupttodesursachen bei Menschen mit Diabetes (Herzinfarkt und Schlaganfall). Auch bei Erreichung der HbA1c-Therapieziele kommt es bekanntermaßen in der Regel zu einem progredienten Diabetesverlauf und häufig auch mit finalen makrovaskulären Endpunkten [ACCORD 2008, Duckworth 2009, ElSayed 2023, Patel 2007].
Wie kann man effizient phänotypisieren?
Die Klassifizierung von Patienten mit Typ-2-Diabetes anhand der klassischen klinischen und Labormarker (HbA1c, Glukose, Lipide, BMI und Blutdruck) ist eine Klassifizierung nach Symptomen und gibt praktisch keinen Einblick in die zugrunde liegenden pathophysiologischen Störungen (Insulinresistenz, ß-Zell-Dysfunktion, Adipogenese und CSI). Bereits vor über 15 Jahren habe ich gemeinsam mit Ko-Autoren das beschriebene Biomarkerkonzept publiziert, das in meiner Praxis von meinen Kollegen und mir seit dieser Zeit auch regelhaft angewendet wird [Pfützner 2008].
Die Beurteilung der ß-Zell-Dysfunktion ist für uns von besonderem Interesse, da immer mehr Medikamente entwickelt wurden, die die ß-Zellen schützen oder ihre Funktionsfähigkeit erhalten sollen, wie z.B. GLP-1-Analoga oder DPPIV-Inhibitoren. Neben den konventionellen Mitteln zur Bewertung der ß-Zellfunktion und der Insulinresistenz (z.B. HOMA-Score oder mahlzeitenbezogene Insulin-/C-Peptid Sekretion), verwenden wir die Bestimmung des intakten Proinsulins (iPI) im Nüchternzustand oder unter Belastung zur Ermittlung des Ausmaßes der ß-Zelldysfunktion und des makrovaskulären Risikos. iPI ist zusammen mit Insulin oder C-Peptid ein Indikator für die insgesamt noch vorhandene Produktionskapazität und Produktionsqualität der ß-Zellen. Es gibt zahlreiche randomisierte, prospektive langjährige Studien mit großen Fallzahlen die belegen, dass iPI nicht nur ein valider Risikoindikator für eine bevorstehende Diabetesmanifestation oder für makrovaskuläre Ereignisse ist [Review in Pfützner 2020], sondern sogar als kardiovaskulärer Risikofaktor angesehen werden muss [Galloway 1992, Nordt 2002]. Wenn Proinsulin am vaskulären Insulinrezeptor bindet, führt dies zur atherogenen Aktivierung der MAP-Kinase im Endothel mit Ausschüttung von atherogenen Entzündungsfaktoren (z.B. Endothelin I) [Forst 2009]. Die supraphysiologische Gabe von intaktem Proinsulin im Rahmen einer früheren pharmazeutischen Produktentwicklung führte unter Anderem zur massiven und unkontrollierten Sekretion von Plasminogen-Aktivator-Inhibitor 1 (PAI-1) aus dem viszeralen Fettgewebe [Nordt 2002, Panalooh 2003]. Dieses Zytokin blockiert bekanntlich die physiologisch notwendige Thrombolyse [Munkvad 1990]. Nachdem es bei der Phase II dieser Medikamentenentwicklung zu mehreren unerklärlichen und teilweise auch fatalen makrovaskulären Ereignissen bei Patienten mit neumanifestem Typ-1- und Typ-2-Diabetes gekommen war, wurde die Entwicklung von Proinsulin als Antidiabetikum sofort eingestellt [Galloway 1992].
Die Bestimmung eines erhöhten Nüchtern iPI bei einem normoglykämischen Patienten ist in jedem Fall hinweisend für eine massive ß-Zelldysfunktion und klinisch relevante Insulinresistenz [Pfützner 2004b], insbesondere auch beim CSI-getriebenem Phänotyp, den man durch Messung von iPI einfach diagnostizieren kann. Differentialdiagnostisch kommen erhöhte Nüchtern-iPI-Werte nämlich ansonsten nur bei einem (sehr seltenen) Proinsulinom [Fadini 2011, Pérez-Pevida 2016] oder in der frühen Manifestationsphase eines Typ-1-Diabetes vor [Bolinder 2005], beides Diagnosen, die man diagnostisch gut abgrenzen kann.
Adiponektin ist ein weiteres physiologisch wichtiges Adipokin, das allerdings im reifen Fettgewebe (also nicht im Präadipozyten) und vom Bindegewebe gebildet wird. Es ist bekannt, dass es die Insulinsensitivität in der Leber und der Peripherie erhöht und gefäßschützende und anti-atherosklerotische Wirkungen hat [Schöndorf 2005, Trujillo 2005]. Bei der viszeralen Adipogenese wird die Adiponektinsekretion unterdrückt, was z.B. zu einer weiteren Zunahme der Insulinresistenz führt [Bastard 2006]. Die Suppression von Adiponektin ist in unserem Konzept ein Indikator für das Ausmaß der anti-Insulinären viszeralen hormonellen Aktivität und der Leitbiomarker beim Insulinresistenz-getriebenen Phänotyp.
Eine detaillierte Analyse der Framingham-Studienkohorte durch Ridker et al. zeigte, dass CRP-Konzentrationen im normnahen Bereich (> 10 mg/dl) eine unabhängige Stratifizierung des kardiovaskulären Risikos in drei Risikogruppen ermöglichen, wenn sie mit einer hochempfindlichen Testmethode (hsCRP) gemessen werden [Pearson 2003, Ridker 2001 und 2004]. Dieser Marker hat sich weltweit als Biomarker der chronisch-systemischen Inflammation durchgesetzt und ist Bestandteil der Risikobewertungsrichtlinien vieler wissenschaftlicher Fachgesellschaften, darunter der American Heart Association und der American Diabetes Association [Pearson 2003]. Werte unter 1 mg/l beschreiben ein geringes kardiovaskuläres Risiko, 1 – 3 mg/l weisen auf ein moderates kardiovaskuläres Risiko hin, und 3 – 10 mg/l beschreiben eine Bevölkerung mit hohem Risiko. Werte über 10 mg/l können aufgrund anderer unspezifischer Infektionen und Entzündungen auftreten und können daher nicht zur Beurteilung des chronischen systemischen vaskulären Entzündungsprozesses herangezogen werden [Danesh 2004, Pfützner 2006a und 2008]. Beim Typ-2-Diabetes sind erhöhte hsCRP-Werte häufig mit einer deutlichen Insulinresistenz und massiver ß-Zelldysfunktion assoziiert [Pfützner 2006b].
"Blutzuckerkosmetik" vs. phänotypgesteuerte personalisierte Therapie
Die oben geschilderten pathophysiologischen Zusammenhänge lassen bereits erahnen, warum eine ausschließliche therapeutische Fokussierung auf den Blutzucker und den HbA1c unter Umständen keine Verbesserung der makrovaskulären Prognose mit sich bringt. Die therapeutischen Leitlinien weltweit sehen bei Typ-2-Diabetes immer einen Beginn der Therapie mit Diät und Lebensstilmaßnahmen gefolgt vom Einsatz von Metformin als erstem Medikament (First-Line) vor [BÄK 2021, ElSayed 2023, Landgraf 2023]. Begründet wird dies von allen Fachgesellschaften mit der Evidenzlage hinsichtlich der Entwicklung von Spätschäden in vergleichenden Studien. Hierbei wird allerdings außer Acht gelassen, dass es praktisch keine direkten Vergleichsstudien zwischen Metformin und modernen Andidiabetika gibt. Die unspezifischen Insulinsekretagoga (Sulfonylharnstoffe, Glinide) senken vor allem zu Beginn der Therapie äußerst effizient den Blutzucker, beschleunigen aber gleichzeitig die pathophsiologische Progression, und es gibt in der Literatur eine – meines Erachtens nach – mehr als ausreichende Evidenz dafür, dass ihre konsequente Verschreibung und Einnahme durch den Patienten zu einer signifikanten Erhöhung des kardiovaskulären Risikos führt [z.B. Forst 2013, Simpson 2006, Volke 2022]. Sie richten gewissermaßen einen "Zusatzschaden" an, und im direkten Vergleich mit Sulfonylharnstoffen schneidet Metformin daher signifikant besser ab [UKPDS 1998, Johnson 2002].
Dadurch erlangte Metformin trotz seines bekanntermaßen sehr hohen gastrointestinalen Nebenwirkungsprofils den aktuell unangefochtenen Status der "First-Line" Medikation. Obwohl es mit seinem Wirkmechanismus (Hemmung der hepatischen Glukoneogenese aus Fettgewebe) ebenfalls nicht wirklich in die Pathophysiologie der Grundstörungen eingreift, und eigentlich sogar aktiv einer Gewichtsabnahme entgegenwirkt, zeigt es in aktuellen Meta-Analysen einen moderaten Effekt bei der Gewichtsabnahme [Haddad 2023]. In einer der wenigen direkten randomisierten prospektiven monotherapeutischen Head-to-Head Vergleichsstudien gegen ein pathophysiologisch-orientiertes Antidiabetikum (Rosiglitazon) in der ADOPT-Studie [Kahn 2006] war die Metformin-Monotherapie hinsichtlich der Hemmung der Diabetesprogression − gemessen über die Zeit bis zur Notwendigkeit eines zusätzlichen Antidiabetikum − zwar dem Sulfonylharnstoff überlegen, schnitt aber signifikant schlechter ab als Rosiglitazon. Im direkten Vergleich mit Dulaglutide in der AWARD-3 Studie zeigte Metformin ebenfalls schlechtere Therapieergebnisse [Umpierrez 2014]. Es kann daher auch nicht erwartet werden, dass mit Metformin als First-Line Therapie die Progression des Typ-2-Diabetes generell verhindert werden kann.
Zusammenfassend muss an dieser Stelle festgestellt werden, dass Typ-2-Diabetes ein hochkomplexes Krankheitsbild ist, das von mehreren ineinandergreifenden Grundstörungen getragen wird, die in unterschiedlicher Ausprägung vorliegen können. Dies kann zu sehr individuellen klinischen Phänotypen führen, die aktuell alle durch eine standardisierte glukozentrische Therapie eigentlich nicht optimal behandelt werden. Die von mir in diesem Diskussionspapier aufgezeigte − und seit mehr als fünfzehn Jahren praktizierte − Methode einer Phänotypisierung mittels funktioneller Biomarker eröffnet die Möglichkeit einer individuellen und personalisierten Diabetestherapie. Wie wir dies in unserer Praxis machen und welche Ergebnisse wir hierdurch bisher erzielen konnten, möchte ich an gleicher Stelle in einer der nächsten Ausgaben im Teil 2 meines Diskussionspapiers vorstellen.
Literatur beim Verfasser
|
|

Erschienen in: Diabetes-Forum, 2024; 36 (1/2) Seite 32-37