
Seit der Erstzulassung subkutaner Glukosesensoren Ende der 1990er-Jahre hat sich die kontinuierliche Glukosemessung (CGM) von einem ergänzenden Hilfsmittel zu einem zentralen Pfeiler der modernen Diabetestherapie entwickelt. Für Menschen mit Diabetes mellitus, insbesondere für diejenigen, die eine Insulintherapie mit Insulindosisanpassung erhalten, ist eine verlässliche Bestimmung der Glukosekonzentration essenziell.
Zusammenfassung
Die kontinuierliche Glukosemessung (CGM) hat sich in den letzten zehn Jahren von einem ergänzenden Hilfsmittel zu einem zentralen Pfeiler moderner Diabetestherapie entwickelt. Mit steigender Anzahl an Anbietern von CGM-Systemen wächst jedoch die Notwendigkeit für verlässliche Qualitätskriterien: Ungenaue Sensormesswerte können Insulin-Dosieralgorithmen fehlleiten und akute Hypo- oder Hyperglykämien verursachen. In den Vereinigten Staaten (USA) schafft das integrated-CGM-Label (iCGM) der Food and Drug Administration (FDA) seit 2018 Abhilfe, indem es verbindliche Kriterien für Punkt- und Trendgenauigkeit sowie Interferenzprüfungen definiert. Die europäische Medical Device Regulation (EU MDR) verlangt zwar eine Qualitätsbewertung, lässt aber quantitative Vorgaben für die Genauigkeit im Hypoglykämiebereich, Änderungsraten (Rate-of-Change, RoC) der Glukosekonzentration und Langzeitstabilität offen, eine Lücke, die es CE-gekennzeichneten (Conformité Européenne) Systemen erlaubt, mit unzureichendem Qualitätsnachweis in den Markt zu gelangen. Um diese Lücke zeitnah zu schließen, plädiert der vorliegende Beitrag dafür, eine vorliegende iCGM-Zulassung oder eine Studie, die nach dem Guidelineproposal der International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) durchgeführt wurde, als Mindeststandard für Zulassung, Beschaffung und Erstattung in Europa zu übernehmen.
Schlüsselwörter
CGM, kontinuierliche Glukosemessung, Qualitätstandards, iCGM, eCGM
Binding quality standards for CGM: iCGM/eCGM – a first important step
Summary
Continuous glucose monitoring (CGM) has evolved over the past decade from an adjunctive aid to a central pillar of modern diabetes therapy. As the number of CGM manufacturers increases, the need for reliable quality criteria likewise grows: imprecise sensor readings can mislead insulin-dosing algorithms and precipitate acute hypo- or hyperglycemia. In the United States (USA), the integrated CGM (iCGM) label, introduced in 2018, addresses this by setting binding thresholds for point and trend accuracy as well as interference testing. The European Medical Device Regulation (EU MDR) requires quality evaluation, but leaves quantitative requirements for accuracy in the hypoglycemic range, rate-of-change (RoC) accuracy and long-term stability undefined, a gap that allows CE-marked (Conformité Européenne) systems to enter the market with insufficient quality evidence. To close this gap promptly, the present article argues that either an existing FDA (Food and Drug Administration) iCGM authorization or results from a study conducted according to the forthcoming International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) guideline proposal should be adopted without delay as the minimum standard for approval, procurement and reimbursement in Europe.
Keywords
CGM, continuous glucose monitoring, quality standards, iCGM, eCGM
Warum verbindliche Qualitätsstandards für CGM?
Während Systeme zur Glukosemessung in Kapillarblut durch den Nutzer (Self-Monitoring of Blood Glucose, SMBG) Glukose aus kapillärem Vollblut bestimmen und das Ergebnis auf den Plasmaglukosewert umrechnen [Sly 2023], messen CGM-Systeme die Glukose in der interstitiellen Flüssigkeit und überführen den gemessenen Wert mithilfe mathematischer Algorithmen in eine Plasmaglukosekonzentration [Miller 2020]. Die dritte Sensorgeneration brachte bereits werkseitig kalibrierte Sensoren ohne die Notwendigkeit einer Kalibration mittels SMBG hervor. Ursprünglich waren CGM-Sensoren ausschließlich adjuvant zugelassen. Dies bedeutet, dass jede Insulinapplikation weiterhin auf einem bestätigenden SMBG-Wert basieren musste. Mit der nachgewiesenen Genauigkeitssteigerung erteilte die US-amerikanische Zulassungsbehörde FDA (Food and Drug Administration) im Jahr 2016 jedoch erstmals eine Zulassung für den nicht-adjuvanten Einsatz: Therapieentscheidungen dürfen seither, bei Verwendung entsprechend zugelassener Systeme, direkt auf CGM-Messwerten beruhen [Klonoff 2025]. In der EU existiert kein eigener behördlicher Zulassungsstatus namens "non-adjunctive". Maßgeblich ist die in der CE-Kennzeichnung (Conformité Européenne) ausgewiesene Zweckbestimmung. Mehrere Systeme, die in den USA zugelassen bzw. CE-gekennzeichnet sind (z. B. Abbott FreeStyle Libre 3, Dexcom G7) tragen eine Zweckbestimmung, die Therapieentscheidungen auf Basis von CGM-Werten zulässt, vorbehaltlich der in den Gebrauchsanweisungen genannten Ausnahmen, in denen eine bestätigende Blutglukosemessung gefordert ist (z. B. bei Symptomen, die nicht zu den CGM-Werten passen, oder bei Verdacht auf Sensorausfall).
Randomisierte kontrollierte Studien und Daten aus groß angelegten Registern kommen dabei übereinstimmend zu dem Ergebnis, dass der Wechsel von SMBG zu CGM – unabhängig von Alter der Patientinnen und Patienten oder Therapieform – zu signifikanten Verbesserungen bedeutender glykämischer Parameter führt. HbA1c‐Werte sinken, die Rate an Hypoglykämien sinkt sowohl tagsüber als auch nachts, und die gesundheitsbezogene Lebensqualität sowie das psychische Wohlbefinden steigen spürbar [Bolinder 2016, Haak 2017, Yaron 2019, Deshmukh 2020]. Darüber hinaus korreliert die Nutzung von CGM mit deutlichen Rückgängen bei Krankenhauseinweisungen infolge akuter diabetischer Ereignisse, etwa schwerer Hypoglykämien oder diabetischer Ketoazidosen [Martens 2021, Riveline 2024, Guerci 2023], sowie mit einer Reduktion langfristiger kardiovaskulärer und mikrovaskulärer Komplikationen [Eeg-Olofsson 2024, Eeg-Olofsson 2025].
Dieser therapeutische Erfolg ist jedoch direkt abhängig von der Sensorqualität. CGM-Systeme sind dabei längst nicht mehr nur Stand-alone-Geräte. Sie sind in sensorunterstützte Insulinpumpen (Sensor-Augmented Pump, SAP) integriert, welche die CGM-Werte in die Pumpensteuerung einbeziehen. Häufig verfügen sie auch über Schutzfunktionen, wie das Aussetzen der Abgabe bei niedriger Glukosekonzentration. Die Gabe von Mahlzeitenboli erfolgt dabei weiterhin durch den Nutzer selbst. Zudem bilden CGM-Systeme das Herzstück hybrider Closed-Loop-Systeme (Hybrid Closed Loop, HCL), bei denen ein Algorithmus auf Basis der CGM-Daten die Basalabgabe und auch teils Korrekturen automatisiert. Diese Systeme werden unter dem Oberbegriff automatisierte Insulinabgabe (Automated Insulin Delivery, AID) zusammengefasst [van den Boom 2024]. Greifen Regelalgorithmen jedoch auf ungenaue Sensordaten zurück, kann die resultierende Insulindosierung fehlerhaft sein [Klonoff 2025]. Die Folge sind Fehlalarme, inadäquate Bolusgaben und im Extremfall potenziell lebensbedrohliche bzw. fatale metabolische Entgleisungen.
Um Sicherheit, Wirksamkeit und Vergleichbarkeit zu gewährleisten, sollten CGM-Geräte strenge Genauigkeits- und Qualitätsanforderungen erfüllen. In Europa hat sich allerdings gezeigt, dass das bestehende CE-Zertifizierungsverfahren allein nicht ausreicht, um die erforderliche Genauigkeit aller CGM-Systeme sicherzustellen. Nach einem "Der Gewinner erhält alle Ausschreibungen"-Verfahren (Winner-takes-all-Tenders) meldeten Nutzende und medizinisches Fachpersonal aus der Region Kampanien (Italien) im Jahr 2022 beispielsweise erhebliche Genauigkeitsprobleme mit diesem entsprechenden CE-gekennzeichneten Sensor. Die Unter- und Überschätzungen der Glukosekonzentration im Hypo- und Hyperglykämiebereich waren so gravierend, dass das medizinische Fachpersonal das Gerät nicht mehr verordnete und den Betroffenen empfahl, wieder auf SMBG zurückzugreifen [Amendolaria 2023, Caruso 2023]. Ähnliche Warnungen sprach das Diabetes Technology Network (Großbritannien) 2024 für ein HCL-System aus, das mit schweren Hypoglykämien in Verbindung gebracht wurde, für das der Hersteller jedoch keine belastbaren Qualitätsdaten vorlegen konnte [Association of British Clinical Diabetologists 2024].
CGM-Systeme stehen damit an einem kritischen Entwicklungspunkt: Das Angebot auf dem europäischen Markt wächst mit der Zunahme neuer Sensorsysteme internationaler Hersteller rasch. Allerdings erfolgt dies häufig auch trotz sehr begrenzt publizierter Evidenz oder inhomogener Datenlage zur Sensorqualität. Andererseits offenbart die regulatorische Landschaft eine Lücke. Die europäische Medical Device Regulation (MDR, EU 2017/745) verlangt zwar eine Qualitätsbewertung und die Prüfung der Dokumentation durch eine Benannte Stelle, jedoch gibt es derzeit keine Norm, die konkrete Vorgaben für die Genauigkeit von CGM macht [EU 2017]. Die Hersteller können Daten vorlegen, die nach ihrer Auslegung des State of the Art erlangt wurden, und so ein CE-Zeichen erhalten. Da die Prüfer bei den Benannten Stellen für viele Medizinprodukte zuständig sind, sind sie nicht zwingend auf CGM spezialisiert. Ohne klare Vorgaben für Studien, Auswertungen und Akzeptanzkriterien können daher neue Systeme auf den Markt kommen, die sich weder objektiv mit etablierten Produkten vergleichen noch zuverlässig in automatisierte Insulinabgabe-Systeme integrieren lassen.
Seit 2018 gibt die US-amerikanische FDA mit dem Label "Integrated CGM" (iCGM) erstmals einen klar publizierten Genauigkeitsrahmen vor [Klonoff 2025]. Feste Grenzwerte für Punkt- und Trendgenauigkeit sowie Limits für fehlerhafte Trends definieren minimale Qualitätskriterien. 2025 publizierte ein europäisches Gremium klinischer Experten darauf aufbauend die "European CGM Minimum Expectations" (eCGM) [Mathieu 2025]. Diese fordern, die iCGM-Grenzwerte unverändert zu übernehmen und ergänzen weitere Parameter. Seit 2019 erarbeitet eine Arbeitsgruppe (WG-CGM) der International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) Grundlagen für ein verbindliches Studienprotokoll und Akzeptanzkriterien, die die Grundlage für einen weltweiten ISO-Standard darstellen sollen. Dieser soll als harmonisierter ISO-Standard Anwendung im CE-Kennzeichnungsverfahren finden.
Ziel dieses Artikels ist es, übersichtlich darzulegen, weshalb Europa die FDA-iCGM-Zulassung ohne Verzögerung als verbindlichen Mindeststandard nutzen sollte, wie eCGM diese Grenzwerte um die für den EU-Markt essenziellen Anforderungen an Datenoffenheit, Datenschutz und Interoperabilität ergänzt und welchen Weg die IFCC-Initiative einschlägt, um einen eigenständigen, weiter gefassten EN-ISO-Standard zu entwickeln. Dieser greift einige Elemente aus iCGM und eCGM auf, fasst manche Kriterien allerdings enger, verwendet teilweise andere Kriterien und wird durch verbindliche Studienprozeduren, Vergleichs-Standardisierung und klare Berichtspflichten ergänzt.
ANSI: American National Standards Institute
CE: Conformité Européenne (CE-Kennzeichnung)
CEN: Comité Européen de Normalisation (Europäisches Komitee für Normung)
CG-DIVA: Continuous Glucose Deviation Interval and Variability Analysis
CGM: kontinuierliche Glukosemessung
CNX: Contour Next (Gerät zur kapillären Vergleichsmessung)
CV: Variationskoeffizient
DG7: Dexcom G7
DIN: Deutsches Institut für Normung
DGR: Dynamic Glucose Region
DIS: Draft International Standard (Entwurf einer internationalen Norm)
DSGVO: Datenschutz-Grundverordnung
DTS: Diabetes Technology Societye
CGM: European CGM Minimum Expectations
EN-ISO: Europäisch harmonisierte ISO-Norm
FDIS: Final Draft International Standard (Schlussentwurf einer internationalen Norm)
FDA: U. S. Food and Drug Administration
FL3: FreeStyle Libre 3
HCL: Hybrid Closed Loop
IFCC: International Federation of Clinical Chemistry and Laboratory Medicine
INT: Cobas Integra 400 Plus (Laboranalysator)
iCGM: Integrated Continuous Glucose Monitoring
ISO/TC 212: ISO-Technisches Komitee 212 (Klinische Laboruntersuchungen und IVD-Testsysteme)
LCB: Lower Confidence Bound (untere einseitige 95 %-Konfidenzgrenze)
MDR: Medical Device Regulation (EU 2017/745)
MSP: Medtronic Simplera
NWIP: New Work Item Proposal
OJEU: Official Journal of the European Union (Amtsblatt der EU)
RoC: Rate of Change (Änderungsrate)
SAP: Sensor-Augmented Pump (sensorunterstützte Insulinpumpe)
SGB V: Fünftes Buch Sozialgesetzbuch
SMBG: Self-Monitoring of Blood Glucose (Blutzuckerselbstkontrolle)
USA: Vereinigte Staaten von Amerika
YSI: YSI 2300 Stat Plus (Laboranalysator)
CGM-Technologie und klinische Anforderungen
Die meisten heute verfügbaren Sensoren nutzen ein enzymatisch-amperometrisches Messprinzip. Dabei oxidiert Glukose-Oxidase an der Platinelektrode Glukose zu Glucono-δ-Lacton [Guoqiang 2025]. Das entstehende Wasserstoffperoxid wird elektrochemisch weiteroxidiert und erzeugt einen Strom, der bei ausreichend Sauerstoff und abgeschirmt gegen interferierende Substanzen, weitgehend proportional zur Glukosekonzentration ist [Cappon 2019]. Dieses Messverfahren hat jedoch systemimmanente Grenzen. Die Strom-Glukose-Beziehung ist nicht strikt linear, wird aber durch Kalibration und Umrechnungsalgorithmen abgebildet. Praktische Limitierungen entstehen v. a. durch Sauerstofflimitierung, Diffusionsbarrieren, Interferenzen und Alterung des Sensormaterials. Die Sensitivität hängt dabei unter anderem von der verbleibenden Enzymaktivität und der Permeabilität der Membranen ab. Gemessen wird außerdem die interstitielle und nicht die kapilläre Plasmaglukosekonzentration [Morales-Dopico 2024]. Der Kompartimentwechsel bewirkt eine Verzögerung von etwa sechs Minuten, der sich als Time Lag (Zeitverzögerung) in allen Glukoseverläufen niederschlägt [Basu 2013]. Moderne Geräte glätten nicht nur das Rohsignal algorithmisch, um Rauschen zu reduzieren, sondern haben auch prädiktive Algorithmen zur Verkürzung des Time Lags zur kapillären Glukose. Bei CGM-Systemen, die Messwerte in dichter Abfolge liefern, ist der mittlere Time Lag inzwischen kurz: Für Abbott FreeStyle Libre 3 (FL3) beträgt der mittlere Time Lag ca. zwei Minuten [Alva 2023] und für Dexcom G7 (DG7) ca. 3,5 Minuten [Welsh 2024]. Ältere und viele aktuelle CGM-Systeme haben einen Time Lag von etwa fünf bis 15 Minuten. Diese Spannweite ergibt sich aus dem physiologischen Time Lag (~ 6 Minuten) plus einem technischen Time Lag (Diffusion durch Membranen/Beschichtung sowie Signalverarbeitung, Übertragung und Anzeige). Da diese Kennzahl bislang nicht einheitlich ausgewiesen wird, ist der Vergleich zwischen Systemen erschwert. Ein größerer Time Lag führt klinisch dazu, dass z. B. Warnungen vor einer Hypoglykämie erst später erfolgen. Neben der Messgenauigkeit ist die Datenverfügbarkeit entscheidend. Datenlücken bezeichnen Zeitabschnitte, in denen keine verlässlichen CGM-Werte angezeigt oder übertragen werden, etwa durch Signalverarbeitung, Funkunterbrechungen oder temporäre Sensorausfälle. Gerade in dynamischen Phasen können bereits kurze Lücken die rechtzeitige Alarmierung und AID-Funktionen beeinträchtigen.
Beurteilung der Genauigkeit von CGM-Sensoren
Mit steigendem klinischem Einsatz von CGM rückt zwangsläufig die Frage in den Vordergrund, wie sich die Messgenauigkeit eines Sensors rasch und verlässlich charakterisieren lässt. Die gebräuchlichste Kennzahl ist die Mean Absolute Relative Difference (MARD): Der MARD fasst die Abweichung zwischen Sensormesswerten und zeitgleich erhobenen Vergleichsmesswerten im Blut zu einem einzigen Prozentwert zusammen und eignet sich daher für einen einfachen Vergleich verschiedener CGM-Systeme [Bailey 2021]. Seine Einfachheit ist jedoch auch seine größte Schwäche. Erstens basieren generelle Genauigkeitsparameter wie z. B. MARD und Agreement rate nur auf den Sensormesspunkten, für die gleichzeitig eine venöse oder kapilläre Vergleichsmessung vorliegt, wodurch der Großteil der CGM-Daten unberücksichtigt bleibt. Zweitens unterscheiden sie nicht zwischen Über- und Unterschätzungen der Glukosekonzentration, sodass ein systematischer Bias verborgen bleibt. Drittens bilden diese Parameter die Dynamik des Glukoseverlaufs nur dann angemessen ab, wenn die zugrunde liegenden Vergleichsdaten ausreichend Phasen rascher Anstiege oder Abfälle enthalten. Werden solche Phasen im Studiendesign weitgehend vermieden, kann ein günstiger Genauigkeitsparameter resultieren, der die klinisch kritischen Situationen nicht adäquat widerspiegelt. Viertens verdichten diese Genauigkeitsparameter die gesamte Sensorlaufzeit auf Mittelwerte und verschleiern damit einen möglichen Drift über die Tragedauer von bis zu 15 Tagen [Vigersky 2024].
Aktuelle Studien kombinieren den MARD daher mit klinischen Risiko-Metriken wie dem DTS Error Grid und der DTS Trend Accuracy Matrix, die sowohl den Abstand zum Vergleichsmesswert als auch das Risiko falscher Therapieentscheidungen abbilden [Klonoff 2024]. In vielen Studien wird die Änderungsrate (Rate of Change, RoC) – gemessen in mg/dl/min – geringgehalten und selten angegeben [Freckmann 2023]. Die Angabe der RoC ist wichtig, da nur bei einem ausreichenden Anteil höherer RoCs die Funktionalität der Alarme beurteilt werden kann. Eine kürzlich publizierte Studie testete erfolgreich eine klar definierte Studien- und Auswertungsprozedur unter anderem mit einem definierten Anteil von höheren Änderungsraten [Link 2025]. Gerade hier zeigen Sensoralgorithmen ihre Grenzen, ein Umstand, den ein Mittelwert wie der MARD allein nicht aufdeckt.
So verschiebt sich die Qualitätsbewertung moderner CGM-Systeme von einem einzigen Durchschnittswert hin zu einem aus mehreren Metriken bestehen Bewertungskonzept, das statische wie dynamische Aspekte gleichermaßen berücksichtigt.
Klinisch kritische Zonen
Die größten Messunsicherheiten eines CGM-Sensors treten erfahrungsgemäß in drei klinisch besonders sensible Situationen auf.
Hypoglykämer Bereich (< 70 mg/dl bzw. 3,9 mmol/l)
Hypoglykämien können innerhalb von Minuten lebensbedrohend sein. Ein Sensoralarm muss daher nicht nur auslösen, sondern auch rechtzeitig sein. Mit abnehmender Glukosekonzentration sinkt der elektrochemische Strom, das Signal-zu-Rausch-Verhältnis verschlechtert sich, und die Messgenauigkeit nimmt ab [Freckmann 2025]. Das FDA-iCGM-Regelwerk verlangt, dass ≥ 85 % der Sensorwerte höchstens ±15 mg/dl (0,8 mmol/l) vom Vergleichswert abweichen [Klonoff 2025]. Maßgeblich ist allerdings nicht nur der Punktwert, sondern dass die untere einseitige 95 %-Konfidenzgrenze (Lower Confidence Bound, LCB) den Schwellenwert überschreitet. Damit wird statistisch abgesichert, dass die geforderte Mindestleistung auch im ungünstigen Fall gilt. In einer herstellerunabhängigen Studie, in der mehrere CGM-Systeme bei Erwachsenen mit Typ-1-Diabetes verglichen wurden, wurden die Daten nachträglich genau nach diesen iCGM-Sonderkontrollen ausgewertet [Eichenlaub 2025a]. Es kamen drei Vergleichsverfahren zum Einsatz: 2300 Stat Plus (YSI) und Cobas Integra 400 Plus (INT) (beide Laboranalysatoren und FDA-akzeptierte Referenzen) in venösen Proben sowie Contour Next (CNX) (nicht FDA-akzeptiert) in kapillären Proben. Für den Bereich < 70 mg/dl bzw. 3,9 mmol/l (±15 mg/dl bzw. 0,8 mmol/l) ergaben sich folgende Ergebnisse: Abbott FreeStyle Libre 3 (FL3) mit ausreichender Übereinstimmung der Punktwerte von 89,2 – 90,7 % und einer LCB von 84,0 % gegenüber YSI/INT bzw. 86,0 % gegenüber CNX; Dexcom G7 (DG7) mit ausreichender Übereinstimmung der Punktwerte von 80,6 – 81,1 % und einer LCB von 66,7 % bzw. 67,1 %. Da die iCGM-Bewertung auf FDA-akzeptierten venösen Laborreferenzen beruht und die LCB den Schwellenwert ≥85 % überschreiten muss, verfehlte FL3 diese Vorgabe knapp (84,0 %) und DG7 deutlich (67 %) [Eichenlaub 2025a]. Die geringere iCGM-Compliance wird vor allem dem dynamischeren Studienprotokoll (mittlere RoC ≈ 1,3 mg/dl/min gegenüber ≈ 0,8 mg/dl/min) in dieser Studie zugeschrieben. Häufig lagen die Abweichungen der Punktwerte über der Schwelle, die LCB blieb teils stichprobenbedingt knapp darunter. Zugleich betonen die Autoren, dass FL3 und DG7 sichere, genaue Systeme sind, und plädieren für klar spezifizierte Mindestanforderungen (Studiendesign, Datenverteilung, Traceability der Vergleichsmethode) [Eichenlaub 2025a]. Diese Forderung nach Transparenz der Datenverteilung wird durch eine separate CG-DIVA-Analyse (Continuous Glucose Deviation Interval and Variability Analysis) unterstrichen. Bei einem anonymisierten CGM-System lagen im hypoglykämischen Bereich 10 % der Messwerte um mehr als +40 mg/dl (2,2 mmol/l) neben dem Vergleichswert (90-Perzentil) [Eichenlaub 2024b]. Solche Abweichungen können, je nach Richtung, Hypoglykämie-Alarme verzögern oder verfrühen. Im Extremfall bleiben kritische Alarme aus bzw. es kommt zu falschen/frühzeitigen Hypo-Alarmen.
Rasche Konzentrationswechsel
Rasche Glukoseanstiege und -abfälle treten z. B. nach Mahlzeiten und Insulingaben auf. Typischerweise steigen die Werte nach Mahlzeiten mit positiven RoC von +1 mg/dl/min (oder auch höher) an, während sie nach Insulingaben mit negativen RoC häufig bis etwa -3 mg/dl/min fallen. Wegen des physiologischen und technischen Time Lags kann ein CGM-Wert bis zu 15 Minuten hinter dem venösen Vergleichswert zurückliegen [Freckmann 2021]. Um solche Dynamiken systematisch zu prüfen, wurde das Dynamic Glucose Region-Modell (DGR) vorgeschlagen, welches künftig in einen harmonisierten Prüfstandard einfließen soll [Eichenlaub 2024a]. Das DGR definiert dabei zwei "Alarmzonen": "Alert low" umfasst Messpunkte im Bereich ≥70 mg/dl (3,9 mmol/l) und einer Änderungsrate von <-1 mg/dl/min (bzw. so steil, dass binnen 30 Minuten eine Hypoglykämie <70 mg/dl bzw. 3,9 mmol/l erreicht würde); "Alert high" umfasst Werte ≤300mg/dl (16,7 mmol/l), die mit >+1,5 mg/dl/min steigen (bzw. binnen 30 Minuten zu einer Hyperglykämie >250 mg/dl bzw. 13,9 mmol/l führen). Um diese Situationen in Studien angemessen abzubilden, wird empfohlen, dass jeweils ≥7,5 % der Vergleichsmessungen in die Zonen "Alert low" bzw. "Alert high" fallen [Eichenlaub 2024a]. Wie unterschiedlich CGM-Systeme in solch steilen Phasen abschneiden, zeigt eine herstellerunabhängige Studie, in der mehrere CGM-Systeme verglichen wurden [Eichenlaub 2025b]. Im RoC-Bereich >+2 mg/dl/min lag der MARD für die folgenden CGM-Systeme bei 8,1 % für FL3, 8,7 % für DG7 und 20,1 % für Medtronic Simplera (MSP) [Eichenlaub 2025b]. Eine Abweichung von 20 % bedeutet bei 150 mg/dl (8,3 mmol/l) bereits eine Abweichung von ±30 mg/dl (1,7 mmol/l). Solche Diskrepanzen können Trendpfeile verfälschen, automatische Insulinstopps verzögern oder kleine automatische Korrektur-Insulindosen in AID-Systemen fehlsteuern und erhöhen damit das Risiko akuter Hypo- oder Hyperglykämien erheblich. Abweichungen unter 10 % gelten dagegen als klinisch akzeptabel [Freckmann 2023, Eichenlaub 2024a, Eichenlaub 2025b].
Sensorstabilität über die Laufzeit
CGM-Systeme können über Stunden bis Tage einen systematischen Drift aufweisen. Hauptursachen sind die fortschreitende Inaktivierung der Glukose-Oxidase, Protein-/Biofilm-Ablagerungen auf der Membran und elektrochemische Korrosion der Arbeitselektrode [Harris 2013, Wisniewski 2000, Colvin 2013]. Alle drei Prozesse vermindern die Empfindlichkeit der Messung, sodass bei gleicher Glukosekonzentration immer weniger Strom erzeugt wird. Die Sensorstabilität beschreibt dabei, wie konstant der Bias (systematische Abweichung) und die mittlere absolute Abweichung (MARD) über die gesamte Laufzeit bleiben. Parallel dazu müssen Sensoren gegenüber interferierenden Substanzen geprüft sein. Eine vollständige Vermeidung von Interferenzen ist nicht immer möglich. Wo Interferenzen verbleiben, sind deren Bereich und Richtung transparent auszuweisen. Typische interferierenden Substanzen wie Paracetamol [Basu 2016] und Hydroxyurea [Tellez 2021] oxidieren bei nahezu demselben Potenzial wie Wasserstoffperoxid an der Arbeitselektrode und verursachen dadurch einen additiven Strom. Der Sensor zeigt dann fälschlich höhere Messwerte an. Heutige CGM-Systeme messen nicht über die gesamte Laufzeit stabil, was z. B. der sich ändernde MARD zeigt: Dies illustriert eine Studie, in der mehrere CGM-Systeme verglichen wurden [Eichenlaub 2025b]. Unmittelbar nach der Insertion (0 – 12 Stunden) zeigten die CGM-Systeme FL3 und DG7 zur kapillären Vergleichsmethode einen negativen Bias von -5,9 % bzw. -7,8 % und MARD-Werte von 10,9 % bzw. 12,8 %. MSP startete dagegen mit einem Bias von -16,9 % und einem MARD von 20,0 %. Über den gesamten Studienverlauf zuhause gemessen ergeben sich für FL3, DG7 und MSP folgende Werte: Der Bias sank auf -1,7 % bzw. -4,8 % und -13,7 % und der MARD auf 8,7 % bzw. 10,7 % und 15,7 % [Eichenlaub 2025b]. Während FL3 und DG7 nach einer kurzen Einlaufphase eine nahezu konstante Genauigkeit zeigten, blieb MSP über Tage hinweg um mehr als 10 % zu niedrig [Eichenlaub 2025b]. Ein anhaltender Negativ-Bias (z. B. >10 %) führt zu systematisch niedrigeren CGM-Anzeigen und kann Therapieentscheidungen und resultierende Glukosemetriken beeinflussen [Freckmann 2025]. Genau deshalb verlangen iCGM/eCGM Prüfungen einer stabilen Leistungsfähigkeit über die gesamte Tragedauer sowie Transparenz der Qualitätsdaten (Beginn/Mitte/Ende der Laufzeit, Trendbedingungen, Interferenzen).
Regulatorische Landschaft
Wie im vorherigen Kapitel gezeigt, lässt sich die Qualität eines CGM-Systems nur dann verlässlich beurteilen, wenn alle klinisch relevanten Parameter in einheitlich designten Studien erhoben und vergleichbar ausgewiesen werden. Dazu müssen Punkt- und Trendgenauigkeit, das Rate-of-Change-Verhalten, die Interferenzresistenz und die Sensorstabilität in klar definierten Bereichen quantitativ belegt werden. In der Europäischen Union regelt die Medical Device Regulation (MDR, EU 2017/745) die Zulassung von CGM-Systemen [EU 2017]. Die MDR stuft CGM-Systeme als aktive invasive Produkte der Risikoklasse IIb ein. Hersteller müssen daher eine klinische Qualitätsbewertung (Clinical Evaluation Report) der Benannten Stelle vorlegen, die Sicherheits- und Qualitätsdaten mit dem "Stand der Technik" vergleicht. Im aktuellen EU-Regulierungsumfeld agieren Hersteller und Benannte Stellen jedoch ohne verbindliche Vorgaben dazu, welche Studienparameter sie auswählen und welche Qualitätskriterien sie festlegen sollen. Folglich können CE-Entscheidungen auf Datensätzen beruhen, die dynamische Belastungszonen, Traceability der Vergleichsmethode und Stabilität über die gesamte Laufzeit nicht durchgängig abdecken. Dadurch sind Vergleichbarkeit und Validität vor der CE-Kennzeichnung nur eingeschränkt.
Der Hersteller eines CGM-Systems erhält das CE-Zeichen, sobald eine Benannte Stelle das technische Dossier, die Fertigung und das Qualitätsmanagement geprüft und die Konformitätsbewertung positiv abgeschlossen hat. Dies ist jedoch nicht mit einem Nachweis ausreichender Zuverlässigkeit gleichzusetzen. Da keine verbindlichen Standards zu technischen und allgemeinverbindlichen Spezifikationen durch die EU MDR vorgegeben sind, kann der Hersteller selbst festlegen, welche Kriterien Anwendung finden, und eigene Grenzwerte definieren. Auch für Trendgenauigkeit, Interferenzresistenz und Datenlücken nennt die MDR keinerlei Spezifikationen. Zwar fordert die MDR eine klinische Qualitätsbewertung, jedoch bestätigt zunächst der Hersteller eigenverantwortlich die EU-Konformität im Rahmen des Qualitätsmanagementsystems, so dass die Prüfer primär das Verfahren und nicht die reale Performance prüfen [EU 2017]. Eine nicht näher festgelegte "repräsentative Stichprobe" oder der Verweis auf ein als äquivalent deklariertes Referenz-CGM-System genügt. So gelangen auch Systeme mit begrenzter Datenbasis zur CE-Kennzeichnung, sofern sie das Verfahren formal bestehen. Hinzu kommt, dass trotz MDR-Verpflichtung keine europaweit einheitlichen Auslöse- oder Eskalationsschwellen für die Post-Market-Surveillance festgelegt sind, ab denen sicherheitsrelevante Vorkommnisse zwingend Korrekturmaßnahmen nach sich ziehen müssen. Als sicherheitsrelevante Vorkommnisse gelten beispielsweise schwerwiegende Ereignisse im Rahmen der MDR-Vigilanz, systematische Messfehler, ungewöhnlich hohe Fehlalarm- oder Ausfallraten sowie Übertragungsabbrüche mit möglicher Gefährdung.
Einen gänzlich anderen Ansatz verfolgt die US-amerikanische FDA. Seit 2018 ist für die Gerätekategorie "Integrated CGM" (iCGM), die in 21 CFR (Code of Federal Regulations) § 862.1355 kodifiziert ist, die Zulassung von CGM-Systemen an zwölf sogenannte "Special Controls" geknüpft [Klonoff 2025]. Kernstück sind detaillierte Mindestkriterien: Demnach müssen mindestens 85 % aller Messwerte unter 70 mg/dl (3,9 mmol/l) innerhalb von ±15 mg/dl (0,8 mmol/l) liegen, im Zielbereich von 70 – 180 mg/dl (3,9 – 10 mmol/l) müssen ≥ 70 % der Messwerte und oberhalb von 180 mg/dl (10 mmol/l), ≥ 80 % der Messwerte jeweils innerhalb von ±15 % der Messwerte der Vergleichsmessung liegen. Zudem dürfen nicht mehr als 1 % sämtlicher Messwerte eine falsche Trendrichtung anzeigen. Ergänzend verlangt Absatz (b) [Bolinder 2016] eine standardisierte Interferenzprüfung, etwa mit Paracetamol oder Hydroxyurea. Die iCGM-Kriterien stellen damit weltweit die bislang einzigen und zugleich strengsten verbindlichen Standards für die CGM-Genauigkeit dar (Tabelle 1).
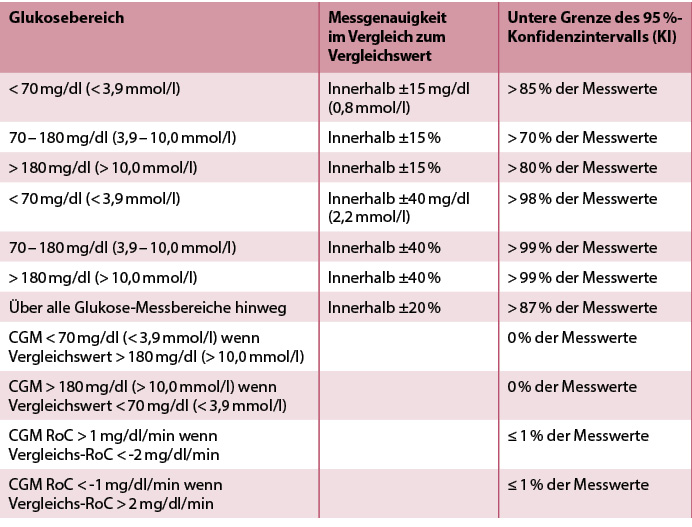 Tab. 1: Vorgeschlagene Mindestleistungsanforderungen für die behördliche Zulassung von CGM-Systemen in Europa [Mathieu 2025].
Tab. 1: Vorgeschlagene Mindestleistungsanforderungen für die behördliche Zulassung von CGM-Systemen in Europa [Mathieu 2025].
Damit verfügt iCGM über die weltweit konkretesten Qualitätskriterien für CGM-Sensoren. Praktisch verbindlich ist der iCGM-Standard allerdings nur in den USA. In Europa kann allerdings dasselbe CGM-System in den Verkehr gebracht werden, ohne dass die Einhaltung dieser Kriterien erneut nachgewiesen werden müsste. Spätere Software- oder Algorithmusänderungen unterliegen allein der Bewertung durch die jeweilige Benannte Stelle. In der fachlichen Diskussion werden die iCGM-Vorgaben daher teils als praktikable Sammlung klar definierter Qualitätskriterien und als Referenzrahmen für europäische Mindestanforderungen herangezogen. Die MDR eröffnet in Artikel 61 ausdrücklich die Möglichkeit, solche internationalen Qualitätskriterien in die Konformitätsbewertung einzubeziehen [EU 2017]. Damit ist juristisch bereits heute der Weg frei, iCGM-Berichte als Ausschluss- oder Auswahlkriterium in Vergabeverfahren und Selektivverträgen zu nutzen. Langfristig sollten die Anforderungen in beiden Rechtsräumen weiterentwickelt und in einem harmonisierten EN/ISO-Standard festgeschrieben werden, damit Qualitätsstudien einheitlich, transparent und ohne methodische Schlupflöcher geplant und bewertet werden.
Warum iCGM jetzt – und wie eCGM hilft
Auf Basis von Artikel 61 der MDR könnte der Zulassungsprozess in Europa ohne Zeitverlust von verbindlichen Qualitätskriterien profitieren, wenn die seit 2018 geltenden iCGM-Grenzwerte der FDA entsprechend übernommen würden [EU 2017]. Diese Kriterien lauten im Kern: <70 mg/dl (3,9 mmol/l): mindestens 85 % der Messwerte innerhalb ±15 mg/dl (0,8 mmol/l) zum Vergleichswert; 70 – 180 mg/dl (3,9 – 10 mmol/l): mindestens 70 % innerhalb ±15 %; >180 mg/dl (10 mmol/l): mindestens 80 % innerhalb ±15 %. Zusätzlich sind höchstens 1 % Trend-Fehlklassifikationen bei schnellen Änderungsraten zulässig. Diese Vorgaben adressieren klinisch relevante Fehlerkonstellationen in Hypo-, Ziel- und Hyperbereichen sowie unter dynamischen Bedingungen [Klonoff 2025]. Das US-Regelwerk lässt jedoch offen, wie ein Hersteller ihre Einhaltung in Europa belegen soll, und adressiert relevante Querschnittsthemen wie Datenschutz, Cybersicherheit und Interoperabilität nicht. Europäische Anforderungen, z. B. Datenschutz-Grundverordnung (DSGVO)-konforme Datenverarbeitung und interoperable Datenaustauschformate bleiben unberücksichtigt. Unter Interoperabilität ist insbesondere die Kompatibilität mit Insulinpumpen und AID/HCL-Systemen sowie mit mobilen Endgeräten zu verstehen. Genau hier setzt der im Jahr 2025 publizierte fachgesellschaftliche Vorschlag der European CGM Minimum Expectations (eCGM) an [Mathieu 2025]. Das internationale Autorenteam um Mathieu et al. behält in seinem Vorschlag zwar die iCGM-Kriterien exakt bei, fordert jedoch ergänzend ein europäisches Prüfprotokoll. Demnach ist zusätzlich zu den iCGM-Vorgaben jede zugelassene Applikationsstelle des Sensors einzubeziehen, die Sensorleistung muss zu Beginn, in der Mitte und am Ende der Tragedauer nachgewiesen werden, mindestens drei Produktionschargen sind separat zu prüfen, Interferenztests müssen sämtliche in der Zielpopulation erwartbaren Substanzen abdecken und das System muss über konstruktive Schutzmechanismen verfügen, die eine Nutzung der Einwegsensoren über die angegebene Tragedauer hinaus technisch verhindern (Abbildung 1). Zudem verpflichtet eCGM die Hersteller, alle klinischen Rohdaten offen zugänglich zu machen, die Konformität ihrer Cyber-Security explizit nach DSGVO-Maßstab nachzuweisen und einen Datenexport nach Norm ISO/IEEE 11073-10425 sicherzustellen [Mathieu 2025].
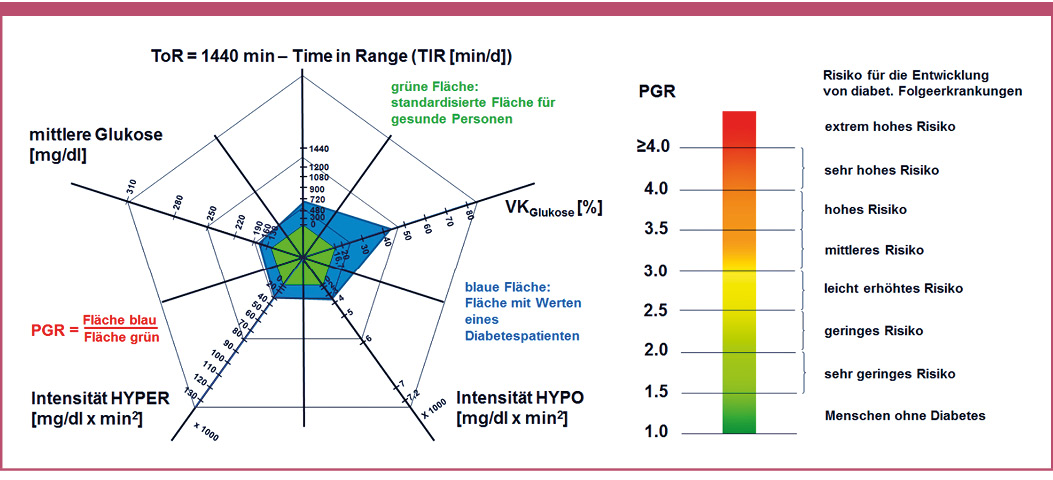 Abb. 1: Mindestanforderungen an Genauigkeit und Leistungsfähigkeit zur Gewährleistung der Sicherheit von Menschen mit Diabetes bei der Anwendung von CGM-Systemen [adaptiert nach Mathieu 2025].
Abb. 1: Mindestanforderungen an Genauigkeit und Leistungsfähigkeit zur Gewährleistung der Sicherheit von Menschen mit Diabetes bei der Anwendung von CGM-Systemen [adaptiert nach Mathieu 2025].
Über die reinen Grenzwerte hinaus legt eCGM besonderen Wert auf die Glaubwürdigkeit der erhobenen Evidenz. Die Qualitätsmetriken müssen in Studien erfüllt werden, die realistische Alltagssituationen abbilden und sämtliche Nutzergruppen einschließen, um ein "hinreichend minimiertes Risiko" für Patientinnen und Patienten zu gewährleisten. Klinische Prüfungen, die qualitätsmindernde Einflussfaktoren wie schnelle Änderungsraten, Extrembereiche, interferierende Substanzen und Datenlücken gezielt ausblenden, liefern keine hinreichend belastbare Evidenz für den Alltag und gelten daher als unzureichend. Vorgeschlagen werden daher unter Praxisbedingungen erhobene Daten zu Sensorlebensdauer, Stabilität, Anwendererfahrungen und Hautverträglichkeit. Für jede Sensor-Applikationsstelle ist eine Mindestanzahl von Vergleichsmessungen von ≥2500 bei Kindern und ≥ 10 000 bei Erwachsenen erforderlich [Mathieu 2025].
Damit verankert das vorgeschlagene eCGM den US-amerikanischen Genauigkeitsmaßstab inhaltlich unverändert und ergänzt diesen um Transparenz-, Datenschutz-/Cybersicherheits- und Interoperabilitätsanforderungen (z. B. Kompatibilität mit Insulinpumpen und AID/HCL-Systemen sowie standardisierte Datenschnittstellen/-exporte). Nicht festgelegt ist jedoch eine verbindliche Studienprozedur. Ohne präzise Vorgaben zu Design, Datenverteilung und Referenzmethode lassen sich die numerischen iCGM-Grenzen prinzipiell erreichen, ohne die reale Qualität unter anspruchsvollen Bedingungen sicherzustellen. In einem begleitenden "Letter to the Editor" weist die "Working Group CGM" (WG-CGM) der IFCC darauf hin, dass Qualitätskriterien ohne ein verbindlich definiertes Studienprotokoll keine echte Vergleichbarkeit schaffen [Pleus 2025]. Die Arbeitsgruppe fordert daher eine gezielte Verteilung der Vergleichsmessungen auf dynamische Glukoseregionen (DGR) mit einem definierten Anteil rascher Glukosewechsel von über 3 mg/dl/min, Prüfungen unter Mahlzeiten- und Aktivitäts-Spitzen sowie eine Prüfpopulation, die zu mindestens 70 – 75 % aus Menschen mit Typ-1-Diabetes besteht. Ebenso seien streng standardisierte Vergleichsmessungen erforderlich. Die Gruppe verweist hierzu auf Daten, die für die Messgenauigkeit der Vergleichsmethode einen Bias <2 % und einen Variationskoeffizenten (CV) <2,5 % fordern [Pleus 2025]. Alle Rohdaten sollen bereits zum Markteintritt offengelegt werden. Die Arbeitsgruppe hat diese Eckpunkte in einen ISO-Initiativantrag eingespeist und strebt gemeinsam mit ISO/TC 212 (Technisches Komitee 212 – Klinische Laboruntersuchungen und In-vitro-Diagnostik-Testsysteme) einen vollständig harmonisierten Standard an. Ein erster Entwurf einer Leitlinie wurde kürzlich durch die IFCC WG-CGM veröffentlicht [Pleus 2026].
Damit ergibt sich für die Praxis ein dreistufiges Modell der Qualitätsforderung: iCGM liefert sofort einen robusten Schutz gegen Systeme mit unzureichender Hypo- oder Trendgenauigkeit, eCGM erweitert diesen Schutz um Rahmenanforderungen (Transparenz-, Datenschutz- und Interoperabilitätskriterien), die in der EU ohnehin gefordert sind, und parallel sollten Ergebnisse aus Studien nach dem in Kürze erscheinenden Konsensus der IFCC WG-CGM zusätzlich zu den FDA-iCGM-Kriterien als Bewertungsbasis herangezogen werden. In Europa kann so die derzeitige Regelungslücke geschlossen werden, ohne Innovation auszubremsen: iCGM schützt sofort vor den größten klinischen Risiken [Klonoff 2025], eCGM adressiert die europäischen Rahmenanforderungen [Mathieu 2025] und die von der IFCC vorangetriebene ISO-Norm sorgt für eine langfristige, globale Harmonisierung [Pleus 2025].
Perspektive und Folgerung – vom iCGM-/eCGM-Regelwerk zur EN-ISO-Norm
Kurzfristiges Ziel: "Safety First"
Solange eine harmonisierte ISO-Norm noch aussteht, sollten die eCGM-Kriterien (bei akzeptablen Studienprozeduren) oder eine FDA-Zulassung als verbindlicher Mindeststandard in nationale Verordnungen, Hilfsmittelverzeichnisse und Beschaffungsrichtlinien aufgenommen werden. Die klinischen Vorteile sind unmittelbar: höhere Genauigkeit im Hypoglykämiebereich, geringere Anteile großer Abweichungen sowie nachgewiesene Robustheit gegenüber interferierenden Substanzen. Zugleich entsteht für Vergabestellen und Kostenträger eine klare, objektive Bewertungsgrundlage für Entscheidungen. Artikel 61 Absatz 10 MDR besagt, dass Benannte Stellen ausdrücklich dazu berechtigt sind, international anerkannte Qualitätskriterien anzuerkennen. Ein iCGM-Bericht erfüllt diese Bedingung vollumfänglich, sofern die zugrunde liegenden Studien nach FDA-anerkannten Verfahren durchgeführt und entsprechend geprüft wurden. Vergabestellen können deshalb auf der Erfüllung der iCGM-Kriterien bestehen und Hersteller, deren CGM-System diese nicht erfüllt, ausschließen. Kostenträger und das Hilfsmittelverzeichnis nach § 139 SGB V (Fünftes Buch Sozialgesetzbuch) sollten denselben Schritt vollziehen. Bereits in den nächsten Ausschreibungszyklen sollte kein europaweites Verfahren mehr Sensoren ohne iCGM-Label oder vergleichbar hochwertige Untersuchung, z. B. IFCC-Studienprozedur [Pleus 2026] als State of the Art, zulassen.
Mittelfristiges Ziel: "Harmonisierte Qualitätskriterien"
Die IFCC WG-CGM erarbeitet derzeit in Abstimmung mit den nationalen Normungsorganisationen DIN (Deutsches Institut für Normung) und ANSI (American National Standards Institute) einen "New Work Item Proposal" (NWIP). Das Dokument soll dem ISO-Technischen Komitee 212 zur Abstimmung vorgelegt werden. Nach dessen Annahme beginnt das reguläre Normungsverfahren mit den Phasen Arbeitsentwurf, Komiteeentwurf, Internationaler Entwurf (Draft International Standard, DIS) und Schlussentwurf (Final Draft International Standard, FDIS). Erfahrungsgemäß dauert dieser Prozess, einschließlich der parallelen Beteiligung des Europäischen Komitees für Normung, mindestens zwei bis vier Jahre. Wird der künftige "ISO/NP CGM-Performance" anschließend als europäische EN-ISO-Norm harmonisiert und im Amtsblatt der EU (Official Journal of the European Union, OJEU) gelistet, entfaltet er europaweit Vermutungswirkung unter der MDR, wonach die Einhaltung der harmonisierten Norm als Konformität mit den einschlägigen grundlegenden Sicherheits- und Qualitätsanforderungen gilt, solange nichts Gegenteiliges nachgewiesen wird. Hersteller erhalten damit klare, einheitlichen Vorgaben für den Marktzugang, während Kliniken, Kostenträger und Benannte Stellen über standardisierte Qualitätskriterien verfügen. Die frühe Übernahme der iCGM-Kriterien in Europa kann diesen Normprozess fachlich stützen, da sie vergleichbare Datensätze erzeugt, die sich an denselben Schwellen und Auswerteprinzipien orientieren (z. B. LCB-Nachweise, Qualitätsnachweise bei hohen Änderungsraten (RoC), Stabilität über die Sensorlaufzeit, Interferenzresistenz). Solche Daten zeigen, was technisch und klinisch praktikabel ist, identifizieren Grauzonen, in denen Präzisierungen nötig sind, und liefern der Normungsarbeit eine belastbare Evidenzbasis über Altersgruppen, Applikationsstellen, Versorgungskontexte und Vergleichsmethoden hinweg. Je mehr robuste Ergebnisse nach einheitlichen Qualitätskriterien vorliegen, desto schneller lassen sich offene methodische Fragen im ISO-Prozess klären und desto zielgenauer können Festlegungen zu Studiendesign, Vergleichsverfahren und Berichtspflichten getroffen werden.
Leitende Botschaft
Alle Stakeholder – Fachgesellschaften, Kostenträger, Benannte Stellen und Industrie – sollten nun auf iCGM-konforme Sensoren umstellen und parallel die eCGM-Erweiterungen (Cyber-Security, Interoperabilität, Offenlegung der Rohdaten) implementieren. Zudem sollten sie sich aktiv an der Entwicklung der ISO-Norm beteiligen, um Transparenz, Zuverlässigkeit und Sicherheit im CGM- und AID-Einsatz zu erhöhen und den Nutzen für Patientinnen und Patienten zu verbessern.
|
|

Erschienen in: Diabetes, Stoffwechsel und Herz, 2026; 35 (1) Seite 26-34