
Insulin hat bekanntermaßen eine einzigartige Stellung unter den zahlreichen Hormonen, die an der Steuerung zellulärer Vorgänge im menschlichen Organismus beteiligt sind. Während die Bedeutung für den Glukosestoffwechsel sogar in der allgemeinen Bevölkerung bekannt ist, sind zahlreiche Wirkungen des Hormons in anderen Bereichen des Körpers selbst in der Ärzteschaft nicht unbedingt präsent. Hierzu gehört sicherlich bereits die vasodilatatorische Funktion des Hormons in den Arteriolen und Kapillaren. Deren Beeinträchtigung ist ein wichtiger, wenn nicht sogar der Hauptfaktor bei der Entstehung der mikrozirkulatorischen Spätschäden bei Diabetes mellitus (z. B. Neuropathie, Nephropathie oder Retinopathie) [Scheede-Bergdahl 2009].
Zusammenfassung
Insulin hat im Körper des Menschen eine einzigartige Stellung als anaboles Hormon im Glukose-, Fett- und Proteinstoffwechsel. Außerdem hat es als Signalhormon wichtige Funktionen im Gefäßsystem und in zahlreichen Organen.Auch das Gehirn ist eines der Zielorgane der Insulinwirkung. Insulinrezeptoren sind auf praktisch allen Zelltypen im zentralen Nervensystem vorhanden, was bestätigt, dass die Insulinsignalübertragung in diesem Organ eine wichtige Rolle spielt. Veränderungen in der Insulinsignalübertragung, die unter anderem durch Insulinresistenz verursacht werden, beschleunigen die Alterung des Gehirns und tragen zur Neurodegeneration mit nachfolgenden neuropsychiatrischen Erkrankungen bei.Das immer bessere Verständnis der Insulinsignalübertragung in verschiedenen Hirnzelltypen/Kreisläufen und wie diese bei Krankheiten verändert wird, hat zu der Hypothese geführt, dass die Behandlung der vaskulären Insulinresistenz durch die pulsatile Insulintherapie (PIT) auch positive Auswirkungen auf neurodegenerative Erkrankungen wie Morbus Alzheimer haben könnte. Bei einzelnen Patienten konnten mittels PIT bereits gute therapeutische Ergebnisse erzielt werden. Wenn sich diese Ergebnisse in klinischen Studien bestätigen lassen, könnte die PIT eine gute Begleittherapie für die pharmazeutische Behandlung der Alzheimer Erkrankung darstellen.
Schlüsselwörter
Alzheimer, Parkinson, Insulinresistenz, pulsatile Insulintherapie
Pulsatile insulin therapy as a treatment option for Alzheimer’s and Parkinson’s disease – pathophysiological background and case study
Summary
Insulin has a unique position in the human body as an anabolic hormone in glucose, fat and protein metabolism. As a signalling hormone, it also has important functions in the vascular system and in numerous organs.The brain is also one of the target organs of insulin action. Insulin receptors are present on practically all cell types in the central nervous system, which confirms that insulin signalling plays an important role in this organ. Alterations in insulin signalling caused by insulin resistance, among other things, accelerate brain aging and contribute to neurodegeneration with subsequent neuropsychiatric disorders.The increasing understanding of insulin signalling in different brain cell types/circuits and how this is altered in diseases has led to the hypothesis that the treatment of vascular insulin resistance by pulsatile insulin therapy (PIT) could also have positive effects on neurodegenerative diseases such as Alzheimer’s disease. Good therapeutic results have already been achieved in individual patients using PIT. If these results can be confirmed in clinical trials, PIT could be a good adjunctive therapy for the pharmaceutical treatment of Alzheimer’s disease.
Keywords
Alzheimer‘s disease, Parkinson‘s disease, insulin resistance, pulsatile insulin therapy
Einleitung
Das Gehirn ist ein weiteres Organ, das in seiner Funktion stark von einer guten Insulinwirkung abhängig ist. Auch wenn das Gehirn mit seinen Neuronen in seiner Funktion nicht so insulinabhängig ist wie z. B. Muskel- oder Fettgewebe, moduliert Insulin die zerebrale Glukoseverwertung und unterstützt damit die Energiegewinnung [Fernandez 2017]. Die Tatsache, dass sich praktisch in allen Zellen im Gehirn Insulinrezeptoren finden lassen, unterstreicht die Bedeutung von Insulin für die Funktionsfähigkeit des Organs [van der Heide 2006]. So ist beispielsweise der Hippocampus die Gehirnregion, die für Gedächtnisbildung und Lernen verantwortlich ist. Hier fördert Insulin die sogenannte synaptische Plastizität, d. h. die Fähigkeit des Gehirns sich durch Erfahrungen zu verändern [van der Heide 2005]. Gleichzeitig schützt Insulin Neuronen vor oxidativem Stress und hemmt schädliche entzündliche Prozesse, die z. B. bei Morbus Alzheimer auftreten, wodurch ein Überleben von Neuronen gefördert wird [Berlanga-Acosta 2020]. Im Hypothalamusbereich, der eine zentrale Rolle bei der Appetitregulation innehat, signalisiert Insulin dem Gehirn, ob genug Energie im Körper vorhanden ist und hilft, das Essverhalten zu steuern [Williams 2001, Kullmann 2020] (s. Abbildung 1).
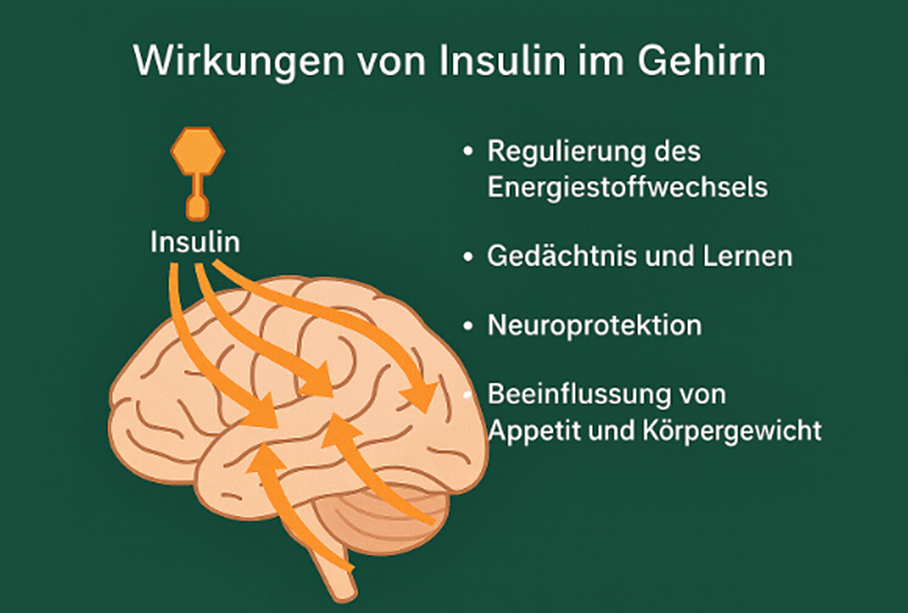 Abb. 1: Bekannte Insulinwirkungen im Gehirn.
Abb. 1: Bekannte Insulinwirkungen im Gehirn.
Insulin-Wirkungen im globalen Stoffwechsel des Gehirns
Das Gehirn hat einen sehr hohen Energiebedarf und ist stark auf einen ausreichend hohen Glukoseumsatz zur Energiegewinnung angewiesen. Des Weiteren ist es das Organ des Körpers, dass den höchsten Cholesteringehalt aufweist (~25 % des Gesamtcholesterins im Organismus). Zerebrales Cholesterin ist nicht nur für die Myelinbildung essenziell, sondern auch für die Funktion der neuronalen Membranen (Chen 2022). Da die Lipoproteine im Plasma die Blut-Hirn-Schranke nicht überwinden können, ist das Gehirn auf seine lokale Cholesterinsynthese für die Myelin- und Synapsenbildung und Optimierung angewiesen. Insulininfusionen bei normalen Glukosewerten führen bei Mäusen zur Expression von Genen, die für die Cholesterin- und Triglyzeridsynthese notwendig sind und zur Suppression der Glykolyse führen, d. h. Insulin führt zu einer Umleitung des neuronalen Stoffwechsels (weg vom Glukosestoffwechsel und hin zum Lipidstoffwechsel) [Cai W 2021, Suzuki 2010, Suzuki 2013]. Während Insulin in den Neuronen also nicht direkt für die Glukoseaufnahme verantwortlich ist, steuert es den essenziellen Substratfluss, der für viele der neuronalen und kognitiven Funktionen des Gehirns verantwortlich ist.
Auf zellulärer Ebene sind Mitochondrien die zentralen Organellen für Energie- und Stoffwechselregulation und eine gestörte mitochondriale Funktion in vielen peripheren Geweben ist mit Diabetes und Insulinresistenz assoziiert [Ruegsegger 2018]. Auch im Gehirn reguliert Insulin die Bildung, Morphologie und Funktion der Mitochondrien [Chen 2022] und ist für die interne zelluläre Reaktion auf Stressoren mitverantwortlich [Konner 2011, Labuèbe 2013]. Da oxidativer Stress eine negative Auswirkung auf die Cholesterinbiosynthese hat, darf an dieser Stelle festgestellt werden, dass die anabolen Effekte von Insulin auf die zerebrale Cholesterinbiosynthese für die normale Gedächtnisfunktion absolut essenziell sind [Chen 2022].
GSK-3β: Glykogensynthase-Kinase-3 beta
MCI: Minor Cognitive Impairment (leichte kognitive Beeinträchtigung)
MMST: Mini-Mental-State-Test
PI3K: Phosphoinositid-3-Kinase
PIT: pulsatile Insulintherapie
Insulinmangel und Insulinresistenz im Gehirn
Es gibt immer mehr Belege dafür, dass Typ-2-Diabetes mit Stimmungsstörungen und neurodegenerativen Erkrankungen unabhängig von der Dysglykämie verbunden ist. So haben Patienten mit Typ-1-Diabetes ähnlich häufig Depressionen wie die Allgemeinbevölkerung [Johnson 2013], während die Prävalenz von Depression bei Typ-2-Diabetes etwa doppelt so hoch ist wie im Bevölkerungsdurchschnitt [Anderson 2001], wobei die Ausprägung der Depression mit dem Schweregrad der Insulinresistenz korreliert [Kan 2013]. Beide Krankheiten beeinflussen sich gegenseitig und die frühe Diagnose einer Depression ist mit einer um 60 % erhöhten Typ-2-Diabetesinzidenz [Mezuk 2008] und schwereren langfristigen diabetischen Komplikationen assoziiert [de Groot 2001]. Die erhöhte Diabetesprävalenz bei Depression lässt sich teilweise aber nicht vollständig durch das erhöhte Typ-2-Diabetesrisiko bei der Einnahme von trizyklischen und noradrenergen Antidepressiva erklären [Khoza 2012, Pan 2012]. Umgekehrt zeigten Menschen mit Typ-2-Diabetes, die mit selektiven Serotonin-Wiederaufnahmehemmern behandelt werden, eine bessere Blutzuckereinstellung [Deuschle 2013, Brieler 2016] und eine erhöhte Insulinempfindlichkeit [McIntyre 2006, Maheux 1997].
Ähnliche Zusammenhänge wurden für die Beziehungen zwischen Diabetes und Angststörungen (z. B. generalisierte Angststörung, Panikstörung, posttraumatische Belastungsstörung und Agoraphobie [Lin 2008, Smith 2013]), Schizophrenie [Kasanin 1926, Mitchell 2013] und anderen neurodegenerativen Erkrankungen (z. B. Demenz [Gudala 2013] oder M. Alzheimer [Arnold 2018, Zhang 2017]) berichtet. Patienten mit Typ-2-Diabetes haben ein etwa 50 % höheres Risiko, eine leichte kognitive Beeinträchtigung oder Alzheimer zu entwickeln [Gudala 2013, Cooper 2015, Zhang 2017]. Postmortale Analysen von Alzheimer-Gehirnen zeigen, dass Signalstörungen von Insulin und dem Insulinartigen Wachstumsfaktor 1 (IGF-1)sowie Alzheimer-typische Ablagerungen von β-Amyloid (Aβ-Plaques) besonders im Schläfenlappen, Hippocampus und Kleinhirn auftreten [Frolich 1998, Talbot 2017]. Ein Rückgang des Insulinrezeptors ( IRS-1) deutet auf eine Insulinresistenz im Gehirn hin [Molony 2010]. Zudem ist die Überaktivierung von wichtigen Proteinen in der neuronalen Signaltransduktionskette (z. B. Phosphoinositid-3-Kinase (PI3K), Proteinkinase B (Akt) oder Glykogensynthase-Kinase-3 beta (GSK-3β)) sowohl im Früh- als auch Spätstadium der Alzheimer-Erkrankung nachweisbar. Kurzgesagt erfolgt physiologischerweise eine Aktivierung von Akt durch PI3K und Akt hemmt wiederum GSK-3β. Wenn dieser Ablauf gestört ist (wie bei Insulinresistenz oder Alzheimer), geraten die neuronalen Zellfunktionen und damit die kognitiven Fähigkeiten aus dem Gleichgewicht [Tramutola 2015]. Alzheimer-Patienten weisen häufig erhöhte Insulinspiegel im Plasma, aber erniedrigte Werte im Liquor auf [Craft 1998, Steen 2015].
Insulinresistenz bei M. Alzheimer
Generell versteht man unter Insulinresistenz ein verringertes Ansprechen von Körpergewebe auf die Insulinwirkung [Goldstein 2002]. Auch im Gehirn lässt sich dieses Phänomen beobachten [Mielke 2005, Tschritter 2006, Sedzikowska 2021], das auf zellulärer Ebene verschiedene Ursachen haben kann (z. B. Reduktion der Anzahl der Insulinrezeptoren, niedrigere Affinität der Rezeptoren oder intrazelluläre Störungen der Signaltransduktionskaskade [Burillo 2021]). Im Gehirn führt die Insulinresistenz zu Verringerung der Neuroplastizität und der Freisetzung von Neurotransmittern an den Neuronen. Somit kann die Insulinresistenz im Gehirn zu Störungen des zellulären Stoffwechsels, gefolgt von kognitiven Problemen und Stimmungsveränderungen beitragen [Arnold 2018]. Folglich ist die Insulinresistenz u. a. auch mit einem Verlust der kognitiven Funktion assoziiert [Willmann 2020].
Die beim Typ-2-Diabetes beobachteten peripheren Stoffwechselphänomene finden sich auch im Gehirn von Alzheimer-Patienten, weshalb bereits mehrfach vorgeschlagen wurde, dass M. Alzheimer als Gehirnvariante des Typ-2-Diabetes oder als Typ-3-Diabetes bezeichnet werden kann [z. B. Hayer 2002, DelaMonte 2008, Burillo 2021]. Gleichzeitig findet sich bei M. Alzheimer mit der pathologischen Anreicherung des sogenannten Tau-Proteins in einigen Gehirnregionen eine krankheitsspezifische histologisch-morphologische Veränderung, die erstmals 1975 im Gehirn von Alzheimer-Patienten nachgewiesen werden konnte [Weingarten 1975]. Insulinresistenz im Gehirn bedeutet zusammengefasst, dass die Neuronen nicht mehr richtig auf Insulin reagieren. Dadurch wird die Aufnahme von Glukose gestört, was zu Energiemangel führt. Gleichzeitig werden wichtige Signalwege wie PI3K-Akt-GSK-3β gestört, was Entzündungen, Zelltod und die Ablagerung von schädlichen Proteinen (wie β-Amyloid und Tau) begünstigt. Langfristig trägt Insulinresistenz so zur Entwicklung kognitiver Beeinträchtigungen und neurodegenerativer Erkrankungen wie Alzheimer bei.
Das Tau-Protein
Das Tau-Protein ist ein mikrotubuli-assoziiertes Protein, das eine wichtige Rolle in den Nervenzellen des Gehirns spielt. Es ist bei gesunden Personen vor allem dafür bekannt, die Stabilität und Struktur von intrazellulären Mikrotubuli zu unterstützen und für den Transport von Nährstoffen und anderen Zellbestandteilen zu sorgen [Sedzikowska 2021]. Es ist wichtig zu beachten, dass Tau für eine normale Gehirnfunktion notwendig ist, da eine Tau-Deletion z. B. mit der Anhäufung von Eisen im Gehirn verbunden ist, was zu Erkrankungen wie der Parkinson-Krankheit führt [Lei 2008]. Tau beeinflusst auch die Synapsen und Kerne von Neuronen [Tramutola 2015], von wo aus es auch in den extrazellulären Raum abgegeben wird [Pooler 2013]. Seine extrazelluläre Funktion ist jedoch nach wie vor unbekannt [Soitropulos 2017]. Die Aktivität des Tau-Proteins wird durch Phosphorylierung moduliert und das Molekül selbst enthält mehr als 85 potenziell phosphorylierte oder phosphorylierbare Serin-, Threonin- und Tyrosinstellen [Sergeant 2008].
Bei M. Alzheimer und anderen sogenannten Tautopathien (z. B. progressive supranukleäre Blickparese (PSP), kortikobasale Degeneration oder frontotemporale Demenz), verändert sich das Tau-Protein durch übermäßige Phosphorylierung und verliert dadurch die Fähigkeit, die Mikrotubuli zu stabilisieren [Arend 2016]. In Gehirnen, die durch Alzheimer charakterisiert sind, weist das Tau-Protein eine dreimal so hohe Hyperphosphorylierung auf wie in normalen Gehirnen [Burillo 2021]. Tatsächlich wurden von 85 phosphorylierbaren Resten mehr als 40 Phosphorylierungsstellen in Tau aus den Gehirnen von Alzheimer-Patienten identifiziert, und 28 Stellen sind ausschließlich phosphoryliert [Rad 2018]. Das veränderte Tau verklumpt zu sogenannten Neurofibrillenbündeln (Tau-Knäuel) im Inneren der Nervenzellen [Brion 2006]. Diese Knäuel tragen zum Absterben der Nervenzellen bei und sind ein typisches Kennzeichen der Alzheimer-Krankheit [Sergeant 2008].
Sowohl die Expression des Tau-Gens als auch die Phosphorylierung des Tau-Proteins selbst werden durch Insulin-Stimulation reguliert [Schubert 2003]. Die GSK-3β ist im Gehirn besonders aktiv und beeinflusst dort Lernen, Gedächtnis und Stimmung. GSK-3β phosphoryliert Tau an mehr als 30 Stellen und könnte daher eine Schlüsselrolle bei der Entwicklung von Alzheimer und neurofibrillären Knäueln spielen [Avila 2012]. Eine gestörte Insulinsignalübertragung kann die Tau-Phosphorylierung durch Erhöhung der GSK-3β-Aktivität verstärken [Schubert 2003]. Eine Aktivierung von GSK-3β erfolgt außerdem durch oxidativen Stress, der als Folge der Insulinresistenz in den Neuronen entstehen kann, und abschließend auch durch einen verringerten Glukosestoffwechsel mit Herabregulierung der O-GlcNAcylierung des Tau-Proteins, d. h. Zuckermoleküle werden von O-glykosidischen Bindungsstellen entfernt, die in der Folge zusätzlich phosphoryliert werden können [Yuzawa 2014]. Dies beeinträchtigt nachweislich die Zellmorphologie und das Zellwachstum sowie den Transport von Organellen, der durch mikrotubuliabhängige Motorproteine vermittelt wird [Burillo 2021]. Die intraneuronale Akkumulation von hyperphosphoryliertem Tau verstärkt den oxidativen Stress und löst Pathologien wie verstärkte Apoptose, mitochondriale Dysfunktion und Nekrose aus [De La Monte 2008].
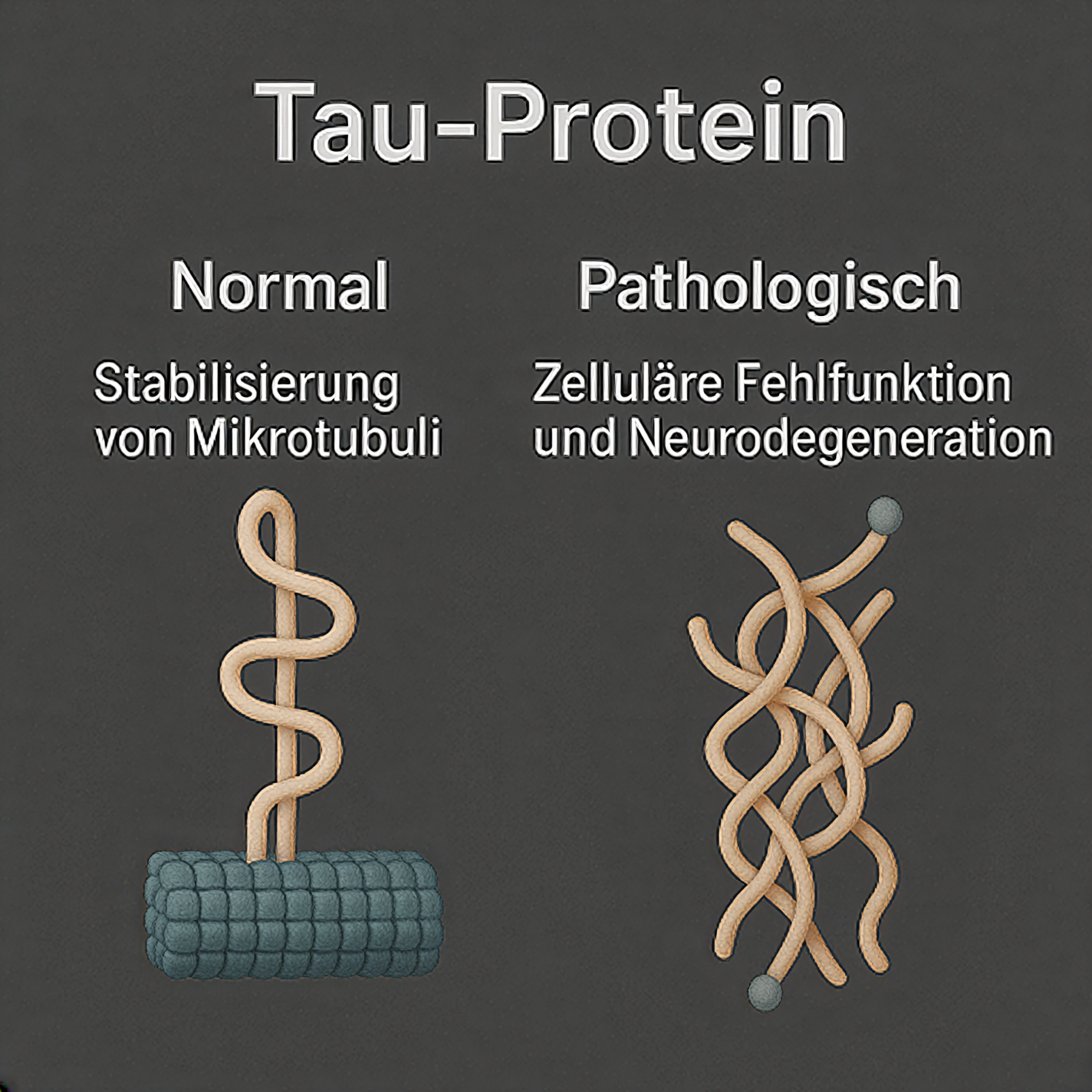 Abb. 2: Funktionelle Unterschiede zwischen normalem und pathologischem Tau-Protein.
Abb. 2: Funktionelle Unterschiede zwischen normalem und pathologischem Tau-Protein.
Tierstudien deuten darauf hin, dass das Tau-Protein die Insulinsignalübertragung im Gehirn regulieren kann, wobei die Deletion von Tau die Reaktion des Hippocampus auf Insulin beeinträchtigt, den Energiestoffwechsel reduziert und zur Anreicherung von Insulin in Form von Oligomeren in hyperphosphorylierten Tau-tragenden Neuronen führt. Der pathophysiologische Verlust der Tau-Funktion dürfte somit mit einer Insulinresistenz des Gehirns verbunden sein und spielt eine Schlüsselrolle bei den kognitiven und metabolischen Beeinträchtigungen von Alzheimer-Patienten [Marciniack 2017, Rodriguez-Rodriguez 2017, Sedzikowska 2021]. Die Anhäufung von Insulin in Neuronen steht in direktem Zusammenhang mit dem Ausmaß der Tau-Hyperphosphorylierung und folgt dem Fortschreiten der Tautopathie; darüber hinaus steht die Anhäufung von Insulin in Zusammenhang mit Insulinresistenz und einer Verringerung der Anzahl der Insulinrezeptoren. Mit dem Fortschreiten der Alzheimer-Krankheitssymptome wird die Tau-Pathologie zunächst im Hirnstamm und im entorhinalen Kortex und später im Hippocampus beobachtet [Jucker 2013].
Zusammengefasst ist die bekannte Pathologie des Tau-Proteins bei M. Alzheimer mit Störungen der Insulinsignaltransduktionswege in praktisch allen Zelltypen des Gehirns mit einer zellulären, vaskulären und metabolischen Insulinresistenz in diesem Organ assoziiert und es liegt auf der Hand, das verschiedene Autoren vorschlagen, M. Alzheimer und andere Tautopathien mit bekannten Interventionen zu behandeln, die auch zur Verbesserung der Insulinresistenz bei Diabetes mellitus eingesetzt werden (Ernährungstherapie, Lifestyle, Insulinsensitizer, Insulin) [Sedzikowska 2021].
Behandlung der Insulinresistenz bei M. Alzheimer
Insulintherapie
Bei Patienten mit Alzheimer-Erkrankung ohne Diabetes kann die klassische periphere Insulinverabreichung eine Hypoglykämie auslösen und ist aufgrund eines gestörten Insulintransports über die Blut-Hirn-Schranke häufig wenig effektiv. Um dieses Problem zu umgehen, wird die intranasale Insulintherapie untersucht, die es ermöglicht, Insulin direkt über perivaskuläre Kanäle des Nervus olfactorius ins Gehirn zu bringen [Kellar 2020]. Erste Studien an kognitiv gesunden Erwachsenen zeigten, dass intranasales Insulin zentrale neuronale Aktivitätsmuster messbar beeinflusst [Kullmann 2016]. Bei Patienten mit leichter kognitiver Beeinträchtigung (Minor Cognitive Impairment, MCI) oder im Frühstadium von Alzheimer, konnte sowohl eine akute als auch eine 21-tägige intranasale Insulinbehandlung das episodische Gedächtnis verbessern [Reger 2008] und die β-Amyloid-Akkumulation modulieren [Craft 2017]. Eine 21-tägige Verabreichung von niedrig- (20 E) und hochdosiertem (40 E) Langzeitinsulin detemir führte bei 60 Patienten zu einer Verbesserung der Gedächtnisleistung, wobei der Effekt bei der höheren Dosis ausgeprägter war [Craft 2017]. Darüber hinaus zeigte eine 120-tägige Therapie bei 104 Teilnehmern mit 20 E oder 40 E Insulin eine signifikante Verbesserung der globalen kognitiven Leistung, gemessen an der Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog12) [Craft 2021]. In zahlreichen weiteren klinischen Studien wurde nachgewiesen, dass intranasales Insulin die Gedächtnisstörungen bei Alzheimer und bei Patienten mit leichtem kognitivem Gedächtnis verbessert. Es wurde festgestellt, dass eine Insulinbehandlung den Abruf von verbalen Informationen nach einer Verzögerung sowie andere kognitive Prozesse wie Orientierung, Urteilsvermögen, soziale Interaktionen und häusliche Aktivitäten verbessert, wie sie von Pflegepersonal bewertet werden [Reger 2008]. Die intranasale Insulinverabreichung könnte somit eine vielversprechende, sichere und nicht-invasive Therapieoption darstellen, um kognitive Funktionen bei MCI und früher Alzheimer-Erkrankung gezielt zu verbessern, insbesondere in höheren Dosierungen [Schmidt 2018, Sedzikowska 2021].
 Abb. 3: Potenzielle Behandlungsmöglichkeiten der Insulinresistenz im Gehirn.
Abb. 3: Potenzielle Behandlungsmöglichkeiten der Insulinresistenz im Gehirn.
Therapie mit Insulinsensitizern
Insulinsensitizer (PPAR-γ-Agonisten bzw. Glitazone) werden eingesetzt, um Gewebe empfindlicher auf niedrigere Insulinspiegel zu machen. Obwohl verschiedene Wirkstoffe dieser Klasse zur Verbesserung der Insulinsensitivität im zentralen Nervensystem getestet wurden, gibt es bislang nur wenige Daten zu ihren Effekten auf das menschliche Gehirn [Kellar 2020]. Ergebnisse einer kleinen Pilotstudie deuten darauf hin, dass PPAR-γ-Agonisten die Insulinsignalübertragung im Gehirn unterstützen und dadurch die kognitive Leistung bei Alzheimer-Patienten stabilisieren oder verbessern könnten [Sato 2011]. In einer Untersuchung an 30 Patienten mit leichter kognitiver Beeinträchtigung oder Alzheimer führte eine sechsmonatige Behandlung mit Rosiglitazon zu einer besseren Gedächtnisleistung [Watson 2005]. Eine Phase-2-Studie zeigte zudem eine kognitive Verbesserung nach 24 Wochen Rosiglitazon-Behandlung [Risner 2006], was in späteren Langzeitstudien aber nicht bestätigt werden konnte [Harrington 2011]. Insgesamt ist noch nicht abschließend geklärt, ob Glitazone die Insulinsensitivität im Gehirn effektiv verbessern können, weshalb ihr therapeutisches Potenzial bei M. Alzheimer noch weiter erforscht werden muss.
Auch wenn für Metformin keine Insulinsensitizer-Wirkung außerhalb der Leber nachgewiesen ist, könnte es das Fortschreiten von Alzheimer verlangsamen oder sogar vorbeugend wirken, aber es gibt noch keine eindeutigen Beweise. Größere, gut kontrollierte klinische Studien laufen derzeit, um die tatsächliche Wirkung besser zu verstehen. Die laufende Phase-2/3-Studie "Metformin in Alzheimer’s Dementia Prevention (MAP)" untersucht die Wirkung von Metformin auf die kognitive Funktion bei Personen mit leichter kognitiver Beeinträchtigung über einen Zeitraum von 18 Monaten [Daly 2025]. Die Ergebnisse werden voraussichtlich 2027 vorliegen.
GLP-1-Rezeptoragonisten wie Liraglutid und Semaglutid wirken nicht nur blutzuckersenkend, sondern haben auch zahlreiche neuroprotektive Eigenschaften (Reduktion von Entzündungen im Gehirn, Verbesserung der Insulinwirkung im zentralen Nervensystem, Schutz vor neuronaler Degeneration, Förderung der Neurogenese und Verbesserung der synaptischen Plastizität [Diz-Chaves 2022, Hölscher 2022]). Diese Effekte könnten helfen, die kognitiven Funktionen bei Alzheimer-Patienten zu erhalten oder zu verbessern. Menschen mit Diabetes, die GLP-1-Agonisten anwendeten, hatten im Vergleich zur Anwendung anderer Antidiabetika ein verringertes Risiko für Demenz [Gejl 2016, Desouza 2015]. Zwei große Phase-3-Studien (EVOKE und EVOKE Plus) testen derzeit Semaglutid bei früher Alzheimer-Erkrankung. Ergebnisse werden 2025 bzw. 2026 erwartet [Cummings 2025].
Behandlung der vaskulären Insulinresistenz durch pulsatile Insulintherapie
Was periphere vaskuläre Insulinrezeptoren sensitiv hält, ist ein Zusammenspiel von normalen Insulinspiegeln, wenig Entzündung, niedrigem oxidativem Stress, gesunder Endothelfunktion und aktiven, nicht blockierten Signalwegen. Ein deutlich unterschätzter Faktor ist hierbei allerdings die sogenannte Pulsatilität der β-Zellen: die β-Zellen gesunder Personen geben Insulin nicht in einem gleichmäßigen Fluss, sondern in Form von regelmäßigen Pulsen ab. Die Zellen oszillieren und werden durch neuronale Faktoren so synchronisiert, dass eine pulsatile Insulinsekretion mit 10 bis 12 Pulsen pro Stunde beobachtet wird [Pørksen 2002, Hellmann 2009a].
Die Pulsatilität hat keinen aktuellen Einfluss auf die Stoffwechselwirkung des Insulins, da mit dem Insulin zeitgleich ein Somatostatin-Peak beobachtet wird und es außerdem phasenverschoben zu einer ebenfalls gepulsten Glukagonsekretion kommt [Hellmann 2009b]. Diese komplexe Pulsatilität, die durch gemeinsame Oszillation und Synchronisation der α-, β-, und δ-Zellen in den Langerhans’schen Inseln entsteht, ist ein Trigger für die Erhaltung der Insulinsensitivität von vaskulären Insulinrezeptoren, die die Produktion von Stickstoffmonoxid (NO) und damit eine Vasodilatation und Vasoprotektion in den peripheren Geweben und Organen bewirken [Pørksen 2002, Hellmann 2009a, Forst 2009].
Die erste und am frühesten zu beobachtende Sekretionsstörung beim Typ-2-Diabetes ist tatsächlich der Verlust der pulsatilen Insulinsekretion [Hellmann 2009b, Wahren 2012]. Die pulsatile Insulinfreisetzung bewahrt jedoch die Sensitivität der vaskulären Insulinrezeptoren und der Verlust dieser Pulsatilität fördert Insulinresistenz und Gefäßschäden (s. Abbildung 4).
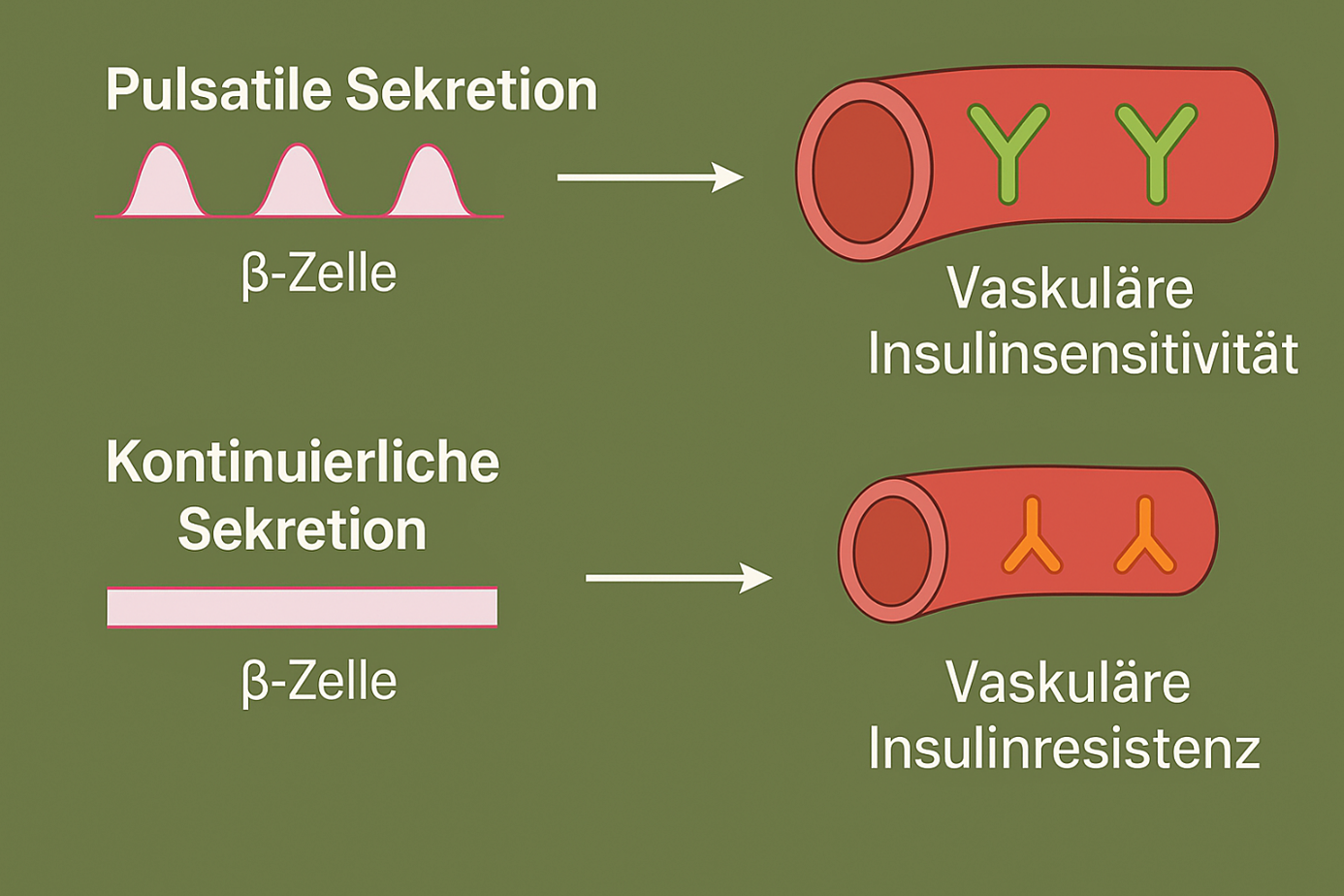 Abb. 4: Kapilläre Vasodilatation und Vasokonstriktion bei pulsatiler bzw. kontinuierlicher pankreatischer Insulinsekretion.
Abb. 4: Kapilläre Vasodilatation und Vasokonstriktion bei pulsatiler bzw. kontinuierlicher pankreatischer Insulinsekretion.
Die Entwicklung von diabetesassoziierten mikrozirkulatorischen Spätschäden selbst bei guter glykämischer Langzeitkontrolle [z. B. Fröhlich-Reiterer 2010, Baz 2013] führte schon vor über 30 Jahren zu Überlegungen, dass eine temporäre Nachahmung der Insulinpulsatilität durch entsprechende intravenöse Insulininfusionen möglicherweise zu Verbesserungen der metabolischen oder vaskulären Insulinresistenz führen könnte [Aoki 2001]. Bereits in den 1980iger Jahren wurde gezeigt, dass die kurzzeitige pulsatile intravenöse Gabe von Insulin zu einer deutlichen Erhöhung der metabolischen Insulinsensitivität führt [Bratusch-Marrein 1986, Paolisso 1988, Heinemann 1989]. Sie wurde daher seit den 90er Jahren unter unterschiedlichen Bezeichnungen und mit unterschiedlichen Behandlungsprotokollen vor allem in den USA und seit ca. fünf Jahren auch wieder in Deutschland zur Behandlung von diabetesassoziierten Spätkomplikationen durch Verbesserung der metabolischen und vaskulären Insulinresistenz eingesetzt. In klinischen Studien konnte gezeigt werden, dass die pulsatile Insulintherapie (PIT) z. B. in der Lage ist, die Progression der diabetischen Nephropathie bei Typ-1-Diabetes zu verlangsamen [Daily 2000, Weinrauch 2007, Weinrauch 2010], bzw. die Nierenfunktion bei Patienten mit Typ-2-Diabetes sogar zu verbessern [Hanna 2020, Manessis 2021]. Neben den renalen Effekten wurden auch positive Auswirkungen der Verbesserung der Insulinresistenz durch eine pulsatile Insulintherapie auf die metabolische Kontrolle, auf die arterielle Hypertonie und andere Diabeteskomplikationen und Komorbiditäten berichtet [Aoki 1993, D’Elia 2011, Elliott 2017, Elliott 2018, Gu 2020, Howard 2021].
Auf der Basis der beobachteten Effekte postulierten wir, dass die pulsatile Insulintherapie durch die temporäre Restaurierung der Signalwirkungen des Insulins an den peripheren Insulinrezeptoren zu einer Verbesserung der metabolischen und der vaskulären Insulinresistenz in praktisch allen Organen führen könnte. Es liegen allerdings nur sehr wenige Informationen bezüglich der praktischen Durchführung der PIT vor, und wir haben daher aus pathophysiologischen Überlegungen heraus ein eigenes PIT-Protokoll mit einer standardisierten Vorgehensweise entwickelt [Hanna 2020, Manessis 2021], um diese Therapie auch bei Patienten mit Alzheimer-Erkrankung im Rahmen von individuellen Heilversuchen einzusetzen.
Fallbeispiel
Die Patientin A. G. (Alter 74 Jahre) leidet seit 2023 an einer diagnostizierten Alzheimer-Demenz mit chronisch progredientem Verlauf. Sie kam Anfang April 2025 für einen Therapieversuch mit ihrem Sohn von einem 500 km entfernten Wohnort in unsere Praxis nach Mainz. Das Erstgespräch musste mit dem Sohn durchgeführt werden, sie wirkte desorientiert und wenig interessiert an der geplanten Therapie. Zur Erfassung ihrer kognitiven Fähigkeiten führten wir einen Mini-Mental-State-Test (MMST) [Folstein 1975] durch. Der Test gibt Aufschluss über die Orientierung (zeitlich und örtlich), das Gedächtnis (Kurzzeit- und Langzeitgedächtnis), die Aufmerksamkeit und Konzentration, Sprache (z. B. Benennung, Nachsprechen, Verstehen), Rechnen (einfache Rechenaufgaben) und visuokonstruktive Fähigkeiten (z. B. Nachzeichnen einer Figur). Das Ergebnis lag mit 10 von 30 möglichen Punkten im Übergangsbereich zwischen mittelschwerer und schwerer Demenz [Crum 1993]. Der ebenfalls durchgeführte DemTec-Screening-Test (DemTect) gibt Aufschluss über Frühstadien kognitiver Störungen, insbesondere MCI und Demenz (z. B. Alzheimer) [Kalbe 2013, Kalbe 2004]. Er erfasst kognitive Bereiche wie z. B. episodisches Gedächtnis, semantisches Gedächtnis, Arbeitsgedächtnis, Wortflüssigkeit, Konzentration und Aufmerksamkeit. Bei diesem Test erreichte die Patientin 7 von 18 Punkten, was ebenfalls auf das Vorliegen einer manifesten Demenz hinweist.
Sie erhielt im Anschluss eine Serie von 10 pulsatilen Insulintherapien (für jeweils 2 Stunden) über einen Zeitraum von drei Wochen. Dies führte u. a. zu einer messbaren Verbesserung der Nierenfunktionswerte (04.04.2025: Kreatinin: 0,82 mg/dl, GFR: 70,4 ml/min – 29.04.2025: Kreatinin: 0,71 mg/dl, GFR: 83,8 ml/min), die wir mangels anderer objektivierbarer Messgrößen als Indikatororgan für eine Verbesserung der Mikrozirkulation im Allgemeinen verwenden. Des Weiteren konnte im Verlauf eine deutliche Verbesserung der Demenzsymptomatik beobachtet werden, die am 29.04.25 auch im MMST (17/30 Punkte = mittelschwere Demenz) und im DemTec (9/18 Punkte = leichte kognitive Beeinträchtigung) Bestätigung fand. Das Abschlussgespräch wurde mit der Patientin im Beisein des Sohnes direkt geführt. Sie ergriff dabei auch spontan das Wort und stellte Fragen zur Therapie. Eine Wiedervorstellung für eine nächste Therapieserie konnte aus logistischen Gründen erst nach vier Wochen erfolgen. Die Patientin zeigte nach anfänglicher Eigenständigkeit wieder zunehmende Anzeichen der Desorientierung und Demenz (Angaben des Sohns). Im Folgenden zeigte sich, dass zumindest in dieser Phase der Erkrankung bei der Patientin eine regelhafte Durchführung der pulsatilen Insulintherapie notwendig war: an den Wochenenden nach Therapiesitzungen war die Patientin orientiert, hatte mehr Energie, kommunizierte klar und verständlich und kam in ihrem Haushalt zeitweise allein zurecht. Konnte in einer Woche keine Therapie gemacht werden, musste sie am Wochenende von den Angehörigen betreut werden und war allein nicht in der Lage, ihren Alltag zu meistern. Diese Beobachtung wurde über mehrere Monate reproduzierbar berichtet, sodass aktuell mit mehreren Kollegen Anstrengungen unternommen werden, ihr die Therapie am Wohnort zur Verfügung zu stellen.
Dieses Ergebnis deckt sich mit den wenigen hierzu vorhandenen Berichten aus der Literatur. Alzheimer-Patienten zeigten auch in früheren Therapieversuchen nach mehreren pulsatilen Insulininfusionen leichte, aber messbare Verbesserungen in Tests wie dem MMST oder Wortlistentests [Morris 2012]. In einzelnen Studien wurde eine verbesserte Konzentrationsfähigkeit und Aufmerksamkeit beschrieben, meist nach mehrwöchiger Behandlung verbunden mit einer durch bildgebende Verfahren (Positronen-Emissions-Tomographie, PET) nachgewiesenen erhöhten Glukoseaufnahme und Steigerung der neuronalen Aktivität [Neth 2017]. Des Weiteren berichten unsere Patienten, die aus anderen Gründen regelmäßig eine pulsatile Insulintherapie erhalten (z. B. wegen Nephropathie oder Neuropathie) über besseren Schlaf und insgesamt mehr Lebensenergie. In Einzelfällen wurde berichtet, dass der kognitive Abbau langsamer voranschreitet, vor allem bei Patienten mit leichtem bis moderatem Alzheimer und Angehörige berichteten von mehr Klarheit im Alltag, besserer Orientierung und teilweise mehr sprachlicher Gewandtheit.
Schlussfolgerungen
Aktuell sprechen vor allem pathophysiologische Überlegungen und wenige Pilotstudien sowie publizierte Fallbeispiele dafür, dass die Behandlung der vaskulären Insulinresistenz mit Hilfe einer regelmäßigen pulsatilen Insulintherapie zur Verbesserung der klinischen Symptomatik bei M. Alzheimer beitragen kann. Diese Ergebnisse sind vielversprechend, allerdings größtenteils aus Pilotstudien oder sind Einzelfallbeobachtungen. Daher sind derzeit keine belastbaren Langzeitdaten verfügbar, und größere, kontrollierte Studien wären erforderlich, um Wirksamkeit und klinische Relevanz abschließend zu bestätigen. Da das Interesse der pharmazeutischen Industrie an dieser mit Normalinsulin durchführbaren Therapie allerdings gering ist, bleibt fraglich, ob es jemals eine Finanzierung für größere, placebo-kontrollierte Studien geben wird, die zur Aufnahme der pulsatilen Insulintherapie in die Therapie-Leitlinien bei Diabetes und Alzheimer notwendig sein dürften.
- Der Verlust der Pulsatilität fördert Endotheldysfunktion und ist ein Treiber diabetischer Mikroangiopathien und evtl. zerebraler Hypoperfusion.
- Die pulsatile intravenöse Gabe von Insulin imitiert die physiologische β-Zell-Rhythmik (~10 – 12 Pulse/Stunde).
- Bei Alzheimer und Parkinson könnte die Verbesserung der vaskulären Insulinresistenz den zerebralen Blutfluss, zellulären Stoffwechsel und damit die Kognition kurzfristig unterstützen.
- Es gibt nur wenig Evidenz: überwiegend Pilotdaten/Einzelfälle, heterogene Protokolle, keine belastbaren randomisierten, kontrollierten Studien mit harten Endpunkten.
- Die PIT ist denkbar bei MCI, früher bis mittlerer Alzheimer-Demenz mit Hinweisen auf vaskuläre Insulinresistenz/Endotheldysfunktion.
- Realistisch gesehen können symptomorientierte Effekte erwartet werden.Um den Erfolg der PIT zu prüfen, können kurze Kognitionstests (MMST/DemTect), alltagsnahe Beobachtungen (Angehörige) und ggf. eGFR/Kreatinin als Mikrozirkulations-Proxy herangezogen werden.
|
|

Erschienen in: Diabetes, Stoffwechsel und Herz, 2025; 34 (6) Seite 336-345