
Der Redner beginnt seinen Vortrag auf der Jahrestagung des "Berufsverbands Deutscher Laborärzte" am 16. Mai 2025 mit dem Hinweis auf das Buch von Guder et al. aus dem Jahr 1999 mit dem Titel: "Proben zwischen Patient und Labor" [Guder 1999]. Hier zitiert er aus dem einführenden Kapitel mit der Überschrift "Traum und Wirklichkeit": "Warum war der Kaliumspiegel erhöht und Glukose normal in der venösen Blutprobe? Antwort: die Probe wurde ohne Abtrennung der Zellen über zwei Stunden in einem nicht belüfteten Auto an einem heißen Tag transportiert. Dies führte dazu, dass die Blutzellen Glukose verbraucht haben und Kalium freigesetzt wurde. Die Konzentration von Kalium ist in den Zellen annähernd 40-Mal höher als im Plasma. Dieser in vitro-Einfluss macht Blut in dem hier betrachteten Aspekt instabil und ungeeignet für eine Glukose-Bestimmung. Kalium kann nur dann zuverlässig gemessen werden, wenn Plasma sofort von den Zellen getrennt wird."
Alle dieser Fehler hätten vermieden werden können, wenn die Vorgaben zu einer geeigneten präanalytischen Handhabung von Blutproben beachtet worden wären. Die Patienten hätten früher am Tag mit weniger Stress untersucht werden können und es wären weniger Kosten entstanden.
Am 19. Februar 2025 erschien im Bundesgesetzblatt die neueste Version der Medizinprodukte-Betreiberverordnung [BMG 2025], in der es in § 10 heißt: "Qualitätssicherungssystem für medizinische Laboratorien, Absatz 1. Wer laboratoriumsmedizinische Untersuchungen durchführt, hat vor Aufnahme dieser Tätigkeit ein Qualitätssicherungssystem nach dem Stand der medizinischen Wissenschaft und Technik zur Aufrechterhaltung der erforderlichen Qualität, Sicherheit und Leistung bei den Anwendern von in-vitro-Diagnostika sowie zur Sicherstellung der Zuverlässigkeit der damit erzielten Ergebnisse einzurichten. Eine ordnungsgemäße Qualitätssicherung nach Satz eins wird vermutet, wenn die Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen in der Fassung vom 30. Mai 2023 (deutsches Ärzteblatt vom 30. Mai 2023, DOI: 10.3238/arztebl.2023.rili_baek_QS_Labor) beachtet wird."
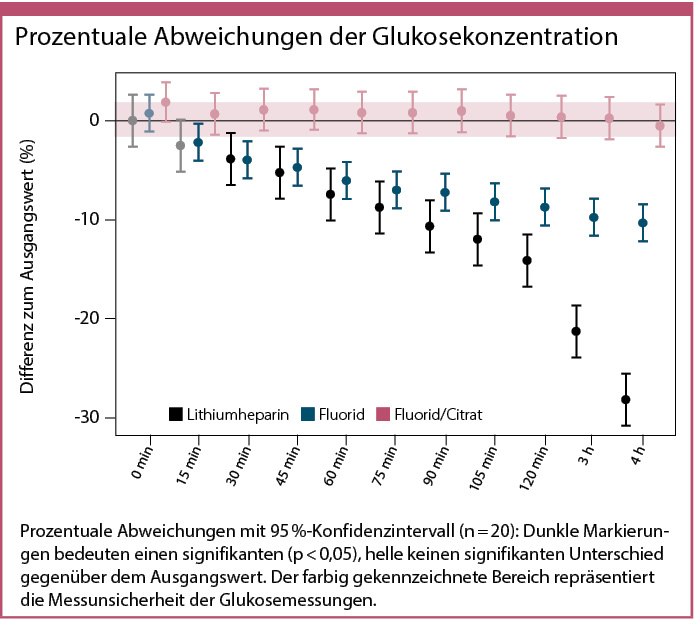 Abb. 1: Daten zur In-vitro-Stabilität der Glukose in unterschiedlichen Untersuchungsmaterialien [mod. nach Petersmann 2022].
Abb. 1: Daten zur In-vitro-Stabilität der Glukose in unterschiedlichen Untersuchungsmaterialien [mod. nach Petersmann 2022].
Der Beginn der Anstrengungen zur Qualitätssicherung in der Laboratoriumsmedizin hat seine formalen Anfänge im Jahr 1971, in der die Richtlinie der Bundesärztekammer zur Durchführung der statistischen Qualitätskontrolle und von Ringversuchen im Bereich der Heilkunde erstmalig publiziert wurde. Seitdem wurde die Rili-BÄK, wie sie üblicherweise abgekürzt wird, immer wieder überarbeitet und weiterentwickelt. Die jüngste Veröffentlichung erschien im Mai 2023 in der Online-Version des Deutschen Ärzteblattes [BÄK 2023]. Seit dem Jahr 2007 ist die Struktur der Rili-BÄK unverändert: sie hat einen allgemeinen Teil A mit grundlegenden Anforderungen an die Qualitätssicherung laboratoriumsmedizinischer Untersuchungen, dem ein Teil B mit fünf Unterteilen folgt, in denen konkrete Vorgaben zur Ergebnisqualität laboratoriumsmedizinischer Untersuchungen definiert werden:
- Quantitative laboratoriumsmedizinische Untersuchungen
- Qualitative laboratoriumsmedizinische Untersuchungen
- Direkter Nachweis und Charakterisierung von Infektionserregern
- Ejakulatuntersuchungen
- Molekulargenetische und zytogenetische laboratoriumsmedizinische Untersuchungen
In den Teilen C und D sind die Zusammensetzungen des Beirats und der Fachgruppen definiert. Im Teil E befinden sich die Anforderungen an die Referenzinstitutionen, während in den Teilen mit den Buchstaben F und G die Übergangsregelungen sowie das Inkrafttreten festgelegt sind.
Im Beirat befinden sich:
- - a) Vertreter der fachlich zuständigen Wissenschaftlichen Fachgesellschaften,
- - b) die Vorsitzenden der Fachgruppen der Teile B,
- - c) ein Vertreter der Bundesärztekammer,
- - d) ein Vertreter der Kassenärztlichen Bundesvereinigung,
- - e) ein Vertreter der Deutschen Krankenhausgesellschaft,
- - f) ein Vertreter des Dachverbandes für Technologen/-innen und Analytiker/-innen in der Medizin Deutschland e. V.,
- - g) ein Vertreter des zuständigen Industrieverbandes,
- - h) drei Vertreter der Länder
- - i) ein Vertreter des Bundesministeriums für Gesundheit,
- - j) ein Vertreter des Bundesinstituts für Arzneimittel und Medizinprodukte und
- - k) ein Vertreter der Physikalisch-Technischen Bundesanstalt.
Dem Beirat gehören als ständige Gäste jeweils ein Vertreter der durch die Bundesärztekammer (BÄK) benannten Referenzinstitutionen gemäß Abschnitt E an.
Die einzelnen Fachgruppen D 1 bis D 5 sind sehr ähnlich dem Beirat zusammengesetzt (Tabelle 1). Bei den Fachgruppen D 3 und D 5 ist zusätzlich das Robert-Koch-Institut vertreten.
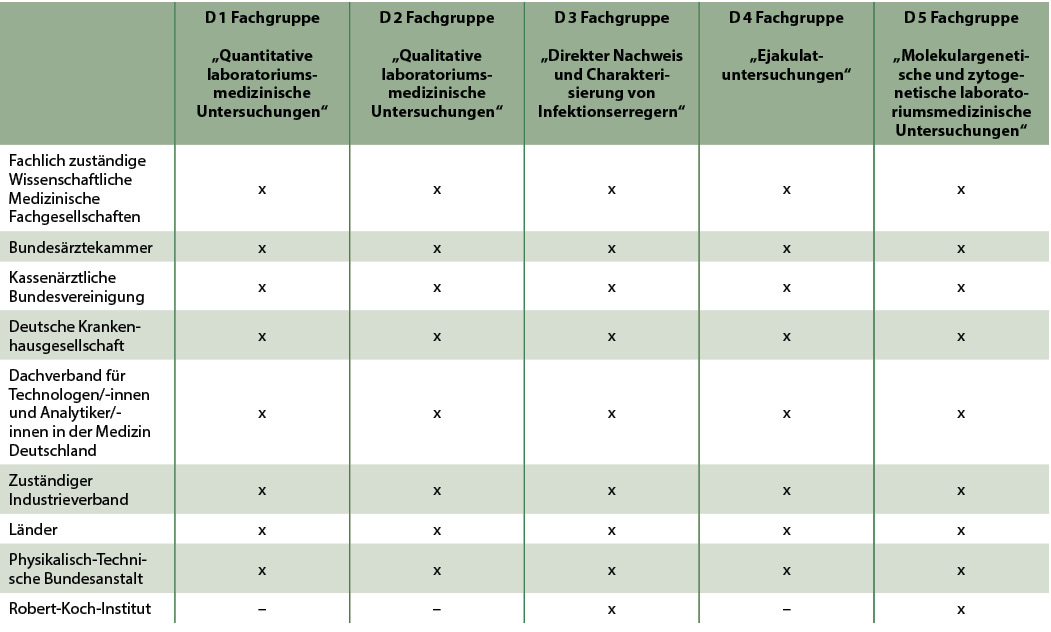 Tab. 1: Zusammensetzung der Fachgruppen D 1 bis D 5. Es sind keine Patientenvertreter in diesen Gremien der BÄK vorgesehen [mod. nach BÄK 2023].
Tab. 1: Zusammensetzung der Fachgruppen D 1 bis D 5. Es sind keine Patientenvertreter in diesen Gremien der BÄK vorgesehen [mod. nach BÄK 2023].
In der Bundesärztekammer werden die Entscheidungen für die Verabschiedung von Veränderungen der Rili-BÄK wie folgt getroffen: die einzelnen Fachgruppen bereiten Änderungsvorschläge vor, die im Beirat besprochen und gegebenenfalls dort als Empfehlung angenommen werden. Diese konsensual erarbeiteten Vorschläge werden anschließend dem Präsidium der Bundesärztekammer zur definitiven Verabschiedung vorgelegt. Es entscheidet also das Präsidium der BÄK über die Veränderungen in der Rili-BÄK. Die Fachgruppen und das Präsidium stellen beratende Gremien dar.
In der Laboratoriumsmedizin werden grundsätzlich drei unterschiedliche Phasen unterschieden:
- die präanalytische Phase
- die analytische Phase
- die postanalytische Phase
Bei der Überarbeitung der Rili-BÄK 2019 wurden Veränderungen im Teil A durchgeführt, wie die Berücksichtigung der allgemeinen Qualitätsmanagement (QM)-Norm des gemeinsamen Bundesausschusses (G-BA), und die Einführung des Peer Reviews als Alternative zum internen Audit. In dieser Version der Rili-BÄK wurden im Teil B 1 die Qualitätsanforderungen an die Messgröße HbA1c für die interne Qualitätssicherung von ± 10 % auf letztlich ± 3 % verschärft. Dies war notwendig geworden, weil sich die Indikationsstellung für das HbA1c in den letzten Jahren prinzipiell geändert hat. Während das HbA1c früher als "Zuckergedächtnis" eingesetzt wurde, ist es seit mehr als zehn Jahren auch als Biomarker für die Diagnose eines Diabetes mellitus zugelassen. Damit wurden die Ansprüche an die analytische Qualität deutlich höher, weil der Graubereich zwischen einem unauffälligen HbA1c-Wert und einem pathologischen Wert recht klein ist, sodass die Messqualität diesen medizinischen Anforderungen entsprechen muss. Darüber hinaus wurden für 30 neue Messgrößen Kriterien für die interne Qualitätssicherung definiert. Wie in den Teilen B 2 und B 3 wurde die Anzahl der Messgrößen mit den Vorgaben für die interne und externe Qualitätssicherung auch im Teil B 1 erhöht. In den anschließenden Teilen C, D und E wurden Redundanzen beseitigt und die Inhalte aktualisiert.
Bei der Überarbeitung der Rili-BÄK 2019 stand damit wie in den vergangenen Jahrzehnten die analytische Phase im Vordergrund. Zu dem Tagesordnungspunkt "überarbeitete Fassung der Rili-BÄK" im Rahmen der vierten Sitzung des Vorstands der Bundesärztekammer am 17./18. Oktober 2019 (Wahlperiode 2019/2023) stehen in dem entsprechenden Protokollauszug folgende Sätze:
"Vorstandsmitglieder [der Bundesärztekammer] äußern den Wunsch, dass die bei dem Messwert HbA1c vollzogene stärkere Berücksichtigung der medizinischen Ansprüche an die Messqualität bei einer nächsten Überarbeitung der Richtlinie auch auf weitere Messwerte ausgedehnt werden soll.
Des Weiteren sollten zukünftig neben der eigentlichen laboratoriumsmedizinischen Untersuchung auch die Anforderungen an die Präanalytik, die z. B. auch den Versand der zu untersuchenden Materialien beinhaltet, überarbeitet werden, da diese die Qualität der Messergebnisse maßgeblich beeinflusst."
Es ist bekannt, dass in der präanalytischen Phase ca. zwei Drittel aller Fehler in der gesamten Prozesskette auftreten, wie z. B. in dem Review von Bonini et al. aus dem Jahr 2002 beschrieben [Bonini 2002].
Vor diesem Hintergrund ist die Präsidiumsentscheidung der Bundesärztekammer zur Fokussierung auf die Präanalytik mehr als gerechtfertigt und aus Sicht der Patientensicherheit ausgesprochen zu begrüßen. Für die Rili-BÄK zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen ist das Thema Präanalytik grundsätzlich nicht neu. So lautet die Zielsetzung der Rili-BÄK unverändert seit 2019:
"Ziel dieser Richtlinie ist, die Qualität laboratoriumsmedizinischer Untersuchungen zu sichern, kontinuierlich zu verbessern und Risiken für Patienten und Anwender so gering wie möglich zu halten. Sie soll insbesondere gewährleisten:
- die Minimierung von Einflussgrößen und Störfaktoren in der Präanalytik,
- die fachgerechte Durchführung der laboratoriumsmedizinischen Untersuchungen einschließlich der Erkennung und Minimierung von Einflussgrößen und Störfaktoren auf die Untersuchungen und
- die korrekte Zuordnung und Dokumentation der Untersuchungsergebnisse einschließlich der Erstellung eines Berichts unter Beachtung von Informationssicherheit und Datenschutz."
Im Teil A der Rili-BÄK findet sich unter dem Kapitel 6 "Laboratoriumsmedizinische Untersuchungen" mit 6.1 ein eigener Abschnitt mit der Überschrift "Präanalytik" [BÄK 2023]:
"6.1.1 Dem Einsender von laboratoriumsmedizinischem Untersuchungsmaterial muss ein für seine Belange relevantes Verzeichnis der vom medizinischen Laboratorium angebotenen laboratoriumsmedizinischen Untersuchungen zur Verfügung gestellt werden.
6.1.2 Eine fachlich kompetente Beratung zur Nutzung des Leistungsangebotes, vor allem hinsichtlich der Auswahl der laboratoriumsmedizinischen Untersuchungen, ggf. des zu wählenden Untersuchungsverfahrens, der Art des Untersuchungsmaterials und der Bewertung der Untersuchungsergebnisse, muss gewährleistet werden.
6.1.3 Aus der Untersuchungsanforderung des Einsenders muss insbesondere folgendes hervorgehen:
- - (1) Identifizierung des Patienten – bei alters- und geschlechtsspezifischen Messgrößen zusätzlich mit Angabe von Geschlecht und Geburtsdatum,
- - (2) die Identifizierung des Einsenders und des Empfängers für die Übersendung des Berichts, wenn er sich vom Einsender unterscheidet,
- - (3) die Art des Untersuchungsmaterials und – falls erforderlich – der anatomische Entnahmeort am Patienten und der Entnahmezeitpunkt,
- - (4) die beauftragten Untersuchungen und
- - (5) die hierfür relevanten klinischen Angaben.
6.1.4 Es müssen Anleitungen für die fachgerechte Entnahme und Behandlung sowie den Transport von Untersuchungsmaterial den Personen zur Verfügung gestellt werden, die hierfür zuständig sind. Die Anleitung für die Gewinnung des Untersuchungsmaterials müssen mindestens enthalten:
(1) die Liste der angebotenen laboratoriumsmedizinischen Untersuchungen oder Verweise darauf,
(2) Anweisungen für
- - (a) die Vorbereitung des Patienten,
- - (b) die Beauftragung der Untersuchung,
- - (c) die erforderlichen Informationen zum Patienten,
- - (d) die Art und Menge des zu entnehmenden Untersuchungsmaterials,
- - (e) die Entnahme von Untersuchungsmaterial mit Beschreibung der Behältnisse für das Untersuchungsmaterial und aller erforderlichen Zusätze,
- - (f) besondere Bedingungen für die Entnahme, die Handhabung, die Lagerung und den Transport des Untersuchungsmaterials falls erforderlich,
- - (g) die unverwechselbare Kennzeichnung des Untersuchungsmaterials und
- - (h) die zu treffenden Maßnahmen zwischen dem Zeitpunkt der Gewinnung und dem Eingang des Untersuchungsmaterials im medizinischen Laboratorium,
(3) Kriterien für die Nachforderung laboratoriumsmedizinischer Untersuchungen,
(4) an Patienten zu übergebende Informationen und Anweisungen hinsichtlich der Vorbereitung und Gewinnung des Untersuchungsmaterials und ggf. Formblätter für die Einverständniserklärung des Patienten zur Gewinnung des Untersuchungsmaterials und für die laboratoriumsmedizinischen Untersuchungen,
(5) Informationen für den Patienten zur Selbstgewinnung eigenen Untersuchungsmaterials sowie für dessen Handhabung, Lagerung und Transport.
6.1.5 Das medizinische Laboratorium muss über eine Anleitung für die Annahme, Kennzeichnung und Bearbeitung von Untersuchungsmaterial für laboratoriumsmedizinische Untersuchungen verfügen.
6.1.6 Kriterien für die Ablehnung von laboratoriumsmedizinischen Untersuchungen sind zu definieren. Erfolgte Ablehnungen sind zu dokumentieren.
6.1.7 Das eingesandte Untersuchungsmaterial und Teilmengen davon müssen eindeutig einem Patienten zuzuordnen sein. Ist dies nicht möglich, darf dieses nicht bearbeitet werden. Der Einsender ist darüber zu informieren. Der Vorgang ist zu dokumentieren.
Wenn das Untersuchungsmaterial einem Patienten nicht zweifelsfrei zuzuordnen ist, aber das Untersuchungsmaterial in gleicher Qualität nicht wiedergewonnen werden kann oder bei kritischem Zustand des Patienten gewonnen wurde, wird nach Rücksprache mit dem Einsender vom medizinischen Laboratorium entschieden, ob die angeforderten laboratoriumsmedizinischen Untersuchungen dennoch durchgeführt werden. Das Ergebnis der Absprache ist zu dokumentieren.
6.1.8 Das medizinische Laboratorium muss bei Eingang des Untersuchungsmaterials prüfen, ob Anhaltspunkte dafür vorliegen, dass
- - (1) die in der Anleitung für die Gewinnung des Untersuchungsmaterials festgelegten Bedingungen nicht eingehalten wurden oder
- - (2) für die angeforderten laboratoriumsmedizinischen Untersuchungen keine zeitgerechte Zustellung erfolgt ist oder
- - (3) die angeforderte Untersuchung nicht durchgeführt werden kann.
Liegen solche Anhaltspunkte vor, muss das medizinische Laboratorium anhand von festgelegten Kriterien entscheiden, ob die Untersuchung dennoch durchgeführt oder neues Untersuchungsmaterial angefordert wird.
Der Vorgang ist zu dokumentieren.
6.1.9 Für zeitkritische laboratoriumsmedizinische Untersuchungen sind ggf. besondere Prozesse zu definieren.
6.1.10 Zur Minimierung von Einflussgrößen und Störfaktoren – nach dem Stand der Wissenschaft und Technik – finden sich Vorgaben in den B-Teilen dieser Richtlinie."
Im Vergleich zur Rili-BÄK des Jahres 2019 wurde bei der Überarbeitung 2023 lediglich der Satz 6.1.10 ergänzt.
Im Teil B 1 wurde bei der Überarbeitung der Rili-BÄK im Jahre 2023 eine neue Tabelle B 1-1 eingeführt (Tabelle 2). In dieser Tabelle befinden sich Vorgaben für die Präanalytik für die beiden Messgrößen Glukose und Kalium.
 Tab. 2: Vorgaben auf zu verwendende Untersuchungsmaterialien* [mod. nach BÄK 2023].
Tab. 2: Vorgaben auf zu verwendende Untersuchungsmaterialien* [mod. nach BÄK 2023].
Glukosebestimmung
Wird Blut einem Patienten abgenommen und befinden sich in dem Blutentnahmeröhrchen keine Zusätze, die den Abbau der Glukose hemmen, sinkt die Glukosekonzentration um knapp 10 % pro Stunde. Die Blutzellen nutzen die vorhandene Glukose, um ihren Energiebedarf zu decken. In der Publikation von Chan et al. aus dem Jahr 1989 in der Zeitschrift Clinical Chemistry wird dieser Zusammenhang eindeutig beschrieben [Chan 1989]. Dabei zeigt diese Untersuchung auch, dass der Versuch, den Glukoseverbrauch nur mit Natriumfluorid zu hemmen erst nach ca. ein bis zwei Stunden erfolgreich ist, sodass auch mit diesem Zusatz die Glukosekonzentration um ca. 10 % unter der in vivo-Konzentration liegt. Ein Kernergebnis einer eigenen Publikation aus dem Jahr 2021 [Fischer 2021] wurde in gekürzter Form im Deutschen Ärzteblatt abgedruckt [Petersmann 2022]. Wie der Abbildung 1 zu entnehmen ist, sinkt bei Blut (nur mit Zusatz Heparinat), das keine Zusätze zur Glykolysehemmung enthält, die Glukosekonzentration nach drei bis vier Stunden um ca. 30 %. Bei Glykolysehemmung mit Natriumfluorid sinken die Glukosekonzentrationen wie erwartet um ca. 10 %. Werden modernere Verfahren zur Glykolysehemmung verwendet, die zusätzlich den pH-Wert der Proben mit Citrat senken, so führt dies zu einer stabilen Glukosekonzentration über vier Stunden und sogar mehrere Tage hinweg [Winter 2016].
In der im Jahr 2023 erschienenen aktualisierten amerikanischen Richtlinie und Empfehlung zur laboratoriumsmedizinischen Analytik im Bereich Diagnosestellung und Management des Diabetes mellitus wird dieselbe Empfehlung zur eingeschränkten Glykolysehemmung der reinen Hemmung mittels Fluorid beschrieben [Sacks 2023].
Dieses Vorgehen wird sowohl seitens der Deutschen Diabetes Gesellschaft (DDG) als auch des Deutschen Zentrums für Diabetesforschung (DZD) mit Nachdruck unterstützt und gefordert.
Kaliumbestimmung
Es ist seit vielen Jahrzehnten bekannt, dass die intrazelluläre Kaliumkonzentration wesentlich höher ist als die extrazelluläre Kaliumkonzentration. Das hat zur Folge, dass die extrazelluläre Kaliumkonzentration in vitro sehr leicht durch eine gestörte Integrität der Zellen beeinflusst wird. Daher ist es im Sinn der Zielsetzung der Rili-BÄK ("Minimierung von Einflussgrößen und Störfaktoren in der Präanalytik") entscheidend, die Integrität der Zellen in der Präanalytik zu erhalten.
In einer deutschen Publikation aus dem Jahr 1998 der damaligen AG Präanalytik der Deutschen Gesellschaft für Klinische Chemie wird Heparin-Plasma bereits klar als das bevorzugte Probenmaterial für die Kaliumbestimmung aufgeführt[Guder 1989]. Die Ursache liegt darin begründet, dass im Rahmen des Gerinnungsvorgangs intrazelluläres Kalium aus Thrombozyten freigesetzt wird. Dieser Zusammenhang wird zum Beispiel in dem Buch von Guder et al. eindrucksvoll gezeigt[Guder 1999]. Die Kaliumkonzentrationen im Serum steigen mit zunehmender Thrombozytenzahl gegenüber der Kaliumkonzentration in Plasma an. Diese Unterschiede können deutlich mehr als 1,5 mmol/l ausmachen und sind klinisch als ausgesprochen relevant einzustufen. Hartland et al. weisen darauf hin, dass Serum ungeeignet ist, die in vivo-Konzentration von Kalium zu bestimmen, was aber das Ziel der Bestimmung der Kaliumkonzentration ist[Hartland 1999]. In einer eigenen Arbeit an gesunden Blutspendern konnten diese Zusammenhänge bestätigt werden[Drogies 2010]. Bei genauerer Betrachtung der Unterschiede in der Kaliumkonzentration zwischen Serum und Plasma fällt auf, dass auch bei grundsätzlich unauffälligen Kaliumkonzentrationen die Werte um mehr als 1,0 mmol/l voneinander abweichen können. Da bei der Bildung von Serum Kaliumionen aus den Thrombozyten freigesetzt werden, handelt es sich hier um einen seit vielen Jahren bekannten Störfaktor, der laut Rili-BÄK minimiert werden muss. Dies ist durch die Vermeidung des Probenmaterials Serum problemlos möglich. Die Kaliumbestimmung muss daher aus Vollblut im Rahmen der Blutgasdiagnostik oder aus dem Probenmaterial Heparin-Plasma erfolgen.
Neben der Medizinprodukte-Betreiberverordnung und der Rili-BÄK ist die In-Vitro-Diagnostika-Richtlinie (IVDR) bei der Durchführung von laboratoriumsmedizinischen Untersuchungen zu beachten [EU 2017]. Ein wichtiges Element sind dabei die "Instructions for Use", oder Gebrauchsanweisungen, die bei CE-markierten Artikeln (Conformité Européenne, Europäische Konformität) verbindlich dazugehören.
In Artikel 10 Absatz 10 (Allgemeine Pflichten der Hersteller) der IVDR findet sich: "Die Hersteller sorgen dafür, dass dem Produkt die Informationen gemäß Anhang I Abschnitt 20 in einer bzw. mehreren Amtssprache(n) der Union beiliegen, die von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegt wird bzw. werden. Die Angaben auf der Kennzeichnung müssen unauslöschlich, gut lesbar und für den vorgesehenen Anwender oder Patienten klar verständlich sein.
Bei Produkten zur Eigenanwendung oder für patientennahe Tests sind die Informationen gemäß Anhang I Abschnitt 20 leicht verständlich und werden in der bzw. den Amtssprache(n) der Union bereitgestellt, die von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegt wird bzw. werden."
Daraus leitet sich die Verpflichtung des Herstellers ab, die notwendigen Informationen zum sicheren Betreiben/Anwenden des Produktes mit Bezug auf die Ausbildung und den Wissenstand des Anwenders bereitzustellen.
Nach dem Produkthaftungsrecht und allgemeinen zivilrechtlichen Grundsätzen gilt: Ein Anwender, der die Gebrauchsanweisung missachtet, handelt möglicherweise grob fahrlässig oder sogar vorsätzlich. Das kann zu Haftungsausschluss oder Mitverschulden führen (z. B. bei Gesundheitsschäden). Die Instructions for Use sind verbindlich im haftungsrechtlichen Sinne.
Das deutsche Medizinprodukterecht-Durchführungsgesetz (MPDG) regelt z. B. in § 83 MPDG die Verantwortung der Leistungserbringer zur Einhaltung der Sicherheit und Zweckbestimmung eines Produkts, was implizit die Berücksichtigung der Packungsbeilage voraussetzt [MPDG 2020].
In Hinblick auf die Kaliumbestimmung geben die Packungsbeilagen wichtige Hinweise auf den Zeitraum, bis wann eine Abtrennung der Zellen zu erfolgen hat, um eine korrekte Kaliumbestimmung durchführen zu können. So findet sich bei:
- DiaSys: "Bis 1 Stunde nach der Blutentnahme von den zellulären Bestandteilen trennen." [DiaSys Diagnostic Systems GmbH 2019]
- Roche: "Kann die Probe nicht innerhalb von 2 Stunden analysiert werden, ist diese von den Zellen zu trennen." [Roche Diagnostics GmbH 2025]
- Siemens: "Serum oder Plasma sollten so bald wie möglich und spätestens nach 2 Stunden nach der Entnahme physikalisch von den Zellen getrennt werden." [Siemens Healthcare Diagnostics Inc. 2019]
- Abaxis Europe: "Durch Venenpunktion erhaltene Vollblutproben sind innerhalb von 60 Minuten nach der Entnahme zu analysieren." [Abaxis Europe 2021]
In einer im Jahr 2025 erschienenen Publikation von Reuter et al. in Plos One [Reuter 2024] wurden die Kaliumbestimmungen aus Serum und Heparin-Plasma bei 201 Probanden verglichen. Dabei wurden einerseits die Probenpaare zeitnah innerhalb von 30 Minuten zentrifugiert, zum anderen wurden weitere Probenpaare erst nach vier, sechs und acht Stunden zentrifugiert. Wie bereits oben beschrieben, sind auch bei dieser Untersuchung die Kaliumkonzentrationen bei dem nach 30 Minuten zentrifugierten Untersuchungsmaterial im Serum erhöht im Vergleich zu den Plasmawerten, da während des Gerinnungsvorgangs Kalium aus den Thrombozyten freigesetzt wird. Diesen Störfaktor gilt es laut Rili-BÄK zu vermeiden. Daher ist für die Kaliumbestimmung grundsätzlich Plasma oder Vollblut im Rahmen der Blutgasanalytik zu verwenden. Die Publikation von Reuter et al. zeigt deutlich einen Anstieg der Kaliumwerte sowohl im Plasma als auch im Serum, wenn die Zentrifugation erst nach vier oder mehr Stunden erfolgt. Die Grundidee, mit der über mehrere Stunden verzögerten Zentrifugation die Realität der laboratoriumsmedizinischen Versorgung abzubilden, wird verfehlt, da – laut IVDR und der entsprechenden Packungsbeilage – die Proben innerhalb von zwei Stunden hätten zentrifugiert werden müssen. Allein aufgrund der Nichtbeachtung der Vorgaben der IVDR mit den Instructions for Use entsprechen alle Messungen, die erst nach einer Zentrifugation von vier oder mehr Stunden erstellt worden sind, nicht dem Stand der Technik. Schlussfolgerungen und Empfehlungen aufgrund dieser – nicht lege artis – erstellten Messwerte haben keine Relevanz. Petersmann et al. konnten in der inzwischen erschienenen Replik auf diese Publikation die Schlussfolgerungen von Reuter et al. anhand der Originaldaten eindeutig widerlegen [Petersmann 2025].
In Hinblick auf die Hämolyse als einem Störfaktor für die Kaliummessung zeigt sich ebenfalls ein ganz einheitliches Bild der untersuchten Packungsbeilagen:
- Abbott: "Für die Bestimmung von Kalium dürfen keine hämolysierten Proben verwendet werden." [Abbott GmbH & Co. KG. 2018]
- DiaSys: "Hämolytische Proben nicht verwenden." [DiaSys Diagnostic Systems GmbH]
- Roche: "Keine hämolysierten Proben verwenden." [Roche Diagnostics GmbH 2025]
- Siemens: "Keine hämolysierten Proben verwenden." [Siemens Healthcare Diagnostics Inc. 2019]
- Abaxis Europe: "Hämolyse kann bei Kalium-Assays zu fälschlicherweise erhöhten Ergebnissen führen." [Abaxis Europe 2021]
Die Zusammenfassung lautet: Hämolytische Proben dürfen gemäß den Packungsbeilagen nicht analysiert bzw. die gemessenen Kaliumwerte nicht in den Befund übernommen werden.
In einem aktuellen Editorial finden sich zahlreiche Hinweise und Belege als Hintergründe für die Überarbeitungen der Rili-BÄK in den vergangenen Jahren[Nauck 2024]. Die Rili-BÄK 2023 selbst wurde Englischsprachig im Journal of Laboratory Medicine publiziert [Ahmad-Nejad 2024]. Die mehr als 50 Autoren zeigen die wertvolle und konstruktive Zusammenarbeit in den unterschiedlichen Gremien der BÄK, die bislang stets konsensuale Beschlüsse gefasst und umgesetzt haben.
Schlussfolgerung
- Die Tabelle B 1-1 in der aktuellen Fassung der Rili-BÄK spezifiziert die präanalytischen Bedingungen für Glukose und Kalium.
- De facto sind in medizinischen Laboratorien mit den bereits länger geltenden Rahmenbedingungen (IVDR mit den Instructions for Use, Medizinprodukte-Betreiberverordnung mit der Forderung nach dem Stand von Wissenschaft und Technik, und Rili-BÄK mit ihren klaren Vorgaben zur Minimierung von Einflussgrößen und Störfaktoren) keine neuen präanalytischen Voraussetzungen für die Messgrößen Glukose und Kalium entstanden.
- Die Rili-BÄK soll die Risiken für Patienten und Anwender so gering wie möglich halten.
- Das Thema Präanalytik wurde in den letzten Jahrzehnten zu wenig beachtet.
- Das Präsidium der BÄK hat den Fokus auf dieses Thema gelenkt.
- Die Medizinprodukte-Betreiberverordnung verlangt den Stand von Wissenschaft und Technik für das Qualitätssicherungssystem.
- Für die Glukosebestimmung geben die Leitlinien seit Jahrzehnten die Verwendung von Plasma – mit Stabilisierung – vor.
- Mithilfe der Zusätze Fluorid und Citrat lässt sich die Glykolyse komplett hemmen, was Grundvoraussetzung für die Messung von korrekten Glukosewerten ist, auch wenn die Analytik erst nach Stunden erfolgt.
- Bei der Bestimmung von Kalium aus Serum kommt eine relevante Störgröße zur Geltung: die Freisetzung von Kalium aus den Thrombozyten während des Gerinnungsvorgangs erhöht die extrazelluläre Kaliumkonzentration und damit die Messwerte. Laut der Zielsetzung der Rili-BÄK ("Minimierung von Einflussgrößen und Störfaktoren in der Präanalytik") gilt es daher, das Probenmaterial Serum zu vermeiden.
- Etliche Packungsbeilagen der In-vitro-Diagnostika-Hersteller weisen für die Bestimmung von Kalium darauf hin, dass die zellulären Bestandteile nach ein bis zwei Stunden abgetrennt werden müssen.
- Hämolytische Proben dürfen – nach Packungsbeilagen – für die Kalium-Analytik nicht verwendet werden.
|
|

Erschienen in: Diabetes, Stoffwechsel und Herz, 2026; 35 (2) Seite 106-113