



Für Patienten mit einem Diabetes mellitus Typ 1 und eine zunehmende Anzahl von Patienten mit einem Diabetes mellitus Typ 2 ist die Insulintherapie lebensnotwendig. Aufgrund der Absorptionskinetik des Insulins im subkutanen Gewebe und der unphysiologischen Verteilung des Insulins im Körper weist die subkutane Insulintherapie einige bedeutende Limitationen auf. Um den physiologischen Insulinbedarf bestmöglich abzubilden, wurden in den vergangenen Jahren zahlreiche neue Insulinformulierungen in die Therapie des Diabetes mellitus eingeführt. Ziel dieses Artikels ist es, die Stärken und Schwächen derzeit verfügbarer und möglicher neuer Insulinformulierungen für die Behandlung des Diabetes mellitus darzustellen.
Einleitung
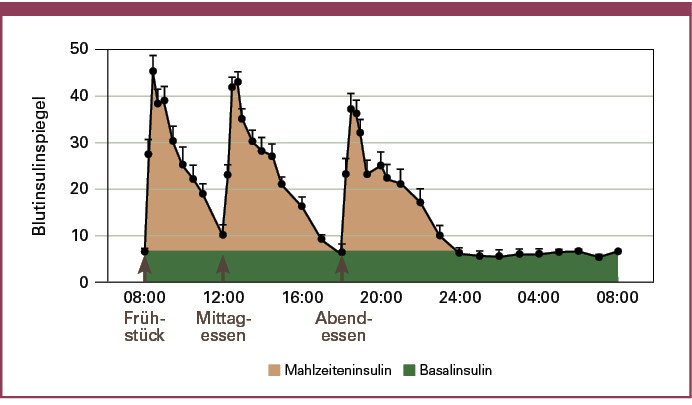
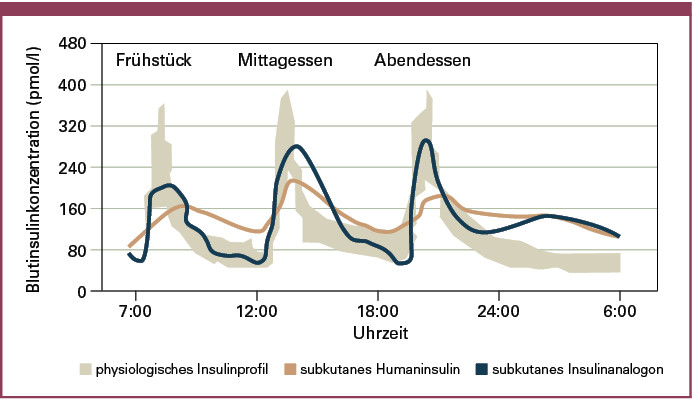
Seit Einführung der Insulintherapie durch Banting und Best im Jahr 1922 ist es das Ziel, eine möglichst physiologische Substitution des Hormons bei Patienten mit einem Diabetes mellitus zu erreichen. Unter funktionellen Aspekten kann hierbei zwischen einer basalen und einer durch Kohlenhydrataufnahme induzierten prandialen Freisetzung des Insulins nach einer Mahlzeit unterschieden werden. Der Insulinbedarf des Körpers in Phasen der Nahrungskarenz, ohne externe Kohlenhydratzufuhr, wird durch die Basalsekretion der Betazelle gewährleistet, während das nach einer Nahrungsaufnahme (postprandial) zusätzlich freigesetzte Insulin ein zu starkes Ansteigen des Blutzuckers nach einer Mahlzeit verhindert. Die enge Kopplung der basalen und prandialen Insulinfreisetzung aus der Betazelle an den aktuellen Blutzuckerspiegel erlaubt beim Gesunden die Stabilisation des Blutzuckers in engen Grenzen (Abbildung 1).
Physiologische Insulinsekretion und subkutane Insulinapplikation
Die Insulinfreisetzung aus der Betazelle unterliegt hierbei nicht nur dem aktuellen Blutzuckerspiegel, sondern wird insbesondere nach einer Mahlzeit von verschiedenen Darmhormonen wie Glucagon-like Peptide 1 (GLP-1) oder Gastric Inhibitory Peptide (GIP) moduliert. Die große Herausforderung an eine externe Insulinsubstitution ist es, ein Insulinprofil zu schaffen, das den aktuellen Insulinbedarf möglichst optimal widerspiegelt. Hierbei stößt die subkutane Insulinapplikation auf schwerwiegende Limitationen. Ein wesentlicher Nachteil ist hierbei in einer vom Blutzucker unabhängigen Wirkung des subkutan applizierten Insulins zu sehen.
Die pharmakokinetischen und pharmakodynamischen Eigenschaften eines subkutan applizierten Insulins werden wesentlich durch die Absorptionskinetik aus dem subkutanen Gewebe und nicht durch den aktuellen Insulinbedarf bestimmt. Hierbei modulieren zahlreiche Faktoren wie die Beschaffenheit des subkutanen Gewebes, die Dichte des lokalen Kapillarbetts, die Hauttemperatur, die lokale Durchblutung u. v. a. die Kinetik der Absorption des Insulins aus dem subkutanen Gewebe und erschweren eine Anpassung des subkutan applizierten Insulins an den tatsächlichen Bedarf. Darüber hinaus kann die Absorption des Insulins aus dem subkutanen Gewebe einer erheblichen intraindividuellen Variabilität unterliegen, was wiederum eine stabile Blutzuckereinstellung erschwert.
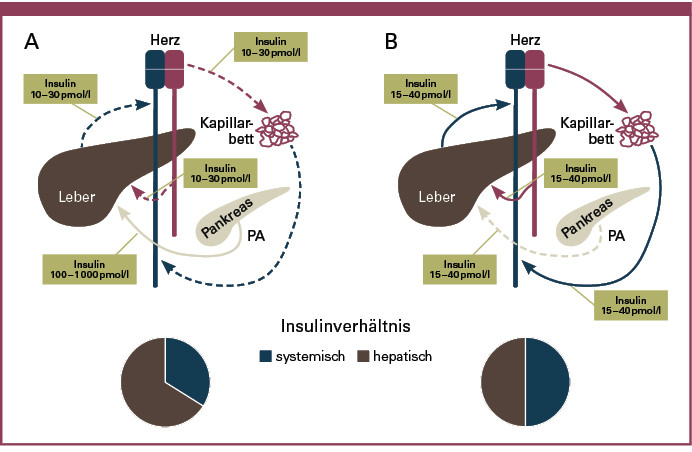
Ein weiterer Nachteil der subkutanen Insulinapplikation ist in der unphysiologischen Insulinverteilung im Körper zu sehen. Nach Sekretion des Insulins in die Pfortader gelangt dieses zunächst in die Leber und bedingt dort eine Hemmung der Glykolyse und der Glukoneogenese. Entgegen diesem physiologischen Weg des Insulins erfolgt die Absorption des Insulins aus dem subkutanen Gewebe überwiegend in die systemische Blutzirkulation (Abbildung 2). Unter physiologischen Bedingungen werden nahezu 40 bis 80 % des portalen Insulins bereits bei der ersten Passage in der Leber abgebaut und nur ein Bruchteil erreicht die peripheren Gewebe. Nach subkutaner Applikation des Insulins erscheint dieses jedoch zunächst in hoher Konzentration in der Peripherie, bevor es sekundär die Leber erreicht.
In diesem Fall fehlt der physiologische Insulingradient zwischen Peripherie und Leber, was mit einer relativen peripheren Hyperinsulinisierung und einem geringeren hepatischen Insulinspiegel einhergeht (1). Diese Verschiebung des peripher-hepatischen Insulingradienten bedingt eine unzureichende Suppression der endogenen Glukosefreisetzung nach einer Mahlzeit und eine überhöhte periphere Glukoseutilisation im Nüchternzustand (2). Der Verlust des peripher-hepatischen Insulingradienten unter einer subkutanen Insulintherapie dürfte wesentlich für das erhöhte Risiko postprandialer Hyperglykämien und nächtlicher Hypoglykämien verantwortlich sein. Darüber hinaus wird die häufig zu beobachtende Gewichtszunahme unter einer subkutanen Insulintherapie mit der peripheren Hyperinsulinisierung in Verbindung gebracht.
Insulintherapie
Von der Einführung der tierischen Insuline (überwiegend Rinder- und Schweineinsulin) über Humaninsulin bis hin zu modernen Insulinanaloga wurden die Insulinformulierungen immer weiter modifiziert mit dem Ziel, die Kinetik der subkutanen Insulinsubstitution dem physiologischen Insulinprofil weiter anzugleichen. Für diesen Zweck stehen heute verschiedene Basal- und Mahlzeiteninsuline sowie die Insulinpumpentherapie zur Verfügung.
Verzögerungsinsuline
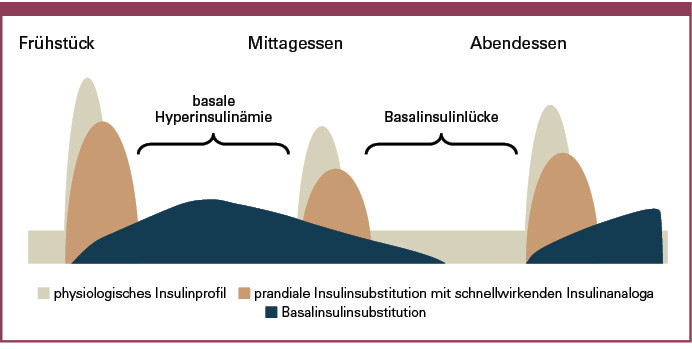
Verzögerungsinsuline zur Abdeckung des basalen Insulinbedarfs sollten eine lange und weitgehend konstante Wirkung aufweisen, um eine Suppression der nächtlichen hepatischen Glukosefreisetzung zu ermöglichen. Ausgeprägte Wirkspitzen sind unerwünscht und erhöhen das Risiko für Hypoglykämien. Neben einer sicheren Abdeckung des basalen Insulinbedarfs in der Nacht für Patienten mit Typ-1- und Typ-2-Diabetes ist für Patienten mit einem Typ-1-Diabetes eine zusätzliche zuverlässige Abdeckung des basalen Insulinbedarfs über den Tag von großer Bedeutung. Insbesondere bei Verwendung einer intensivierten konventionellen Insulintherapie (ICT) mit kurzwirksamen prandialen Insulinanaloga spielt eine optimierte Basalinsulinversorgung über 24 Stunden eine wichtige Rolle, um Hyperinsulinisierung einerseits und Basalinsulinlücken andererseits zu vermeiden. Insuline mit einer zu geringen Halbwertszeit bergen hierbei das Risiko von Hypoglykämien zum Zeitpunkt des Wirkmaximums und von einem Blutzuckeranstieg bei zu frühem Wirkverlust vor erneuter Injektion (Abbildung 3).
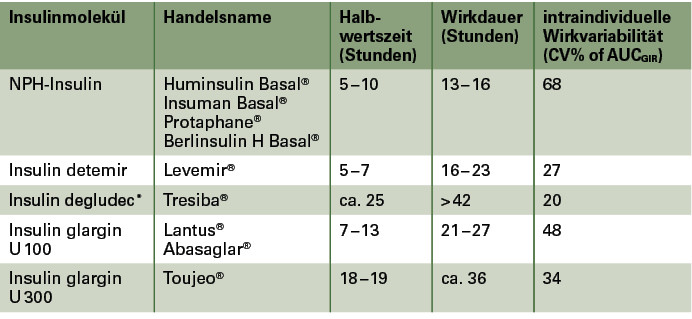
Ein weiteres Problem der Basalinsulinsubstitution ist in einer ungleichförmigen Wirkung und einer hohen intraindividuellen Absorptionsvariabilität verschiedener Basalinsuline zu sehen. Eine präzise Titration eines derartigen Basalinsulins mit hoher Absorptionsvariabilität gestaltet sich oft sehr schwierig und geht mit einem erheblichen Risiko für Hypoglykämien einher. Mit der Entwicklung neuer Basalinsuline sollten Insuline mit einer längeren und gleichmäßigeren Wirkung bei Reduktion der Absorptionsvariabilität geschaffen werden. Der Wirkmechanismus der derzeit zugelassenen Verzögerungsinsuline beruht auf einer Verlangsamung der Insulinabsorption aus dem subkutanen Gewebe und/oder der Bindung an Eiweiße wie Albumin. Hierbei kann eine Kopplung an andere Moleküle wie freie Fettsäuren oder Polyethylenglykol (Insulin detemir [Levemir®], Insulin degludec [Tresiba®] u. a.) oder/und eine Veränderung der Primärstruktur der Insuline (Insulin glargin [Lantus®, Abasaglar®], Insulin detemir [Levemir®], Insulin degludec [Tresiba®]) zum Einsatz kommen. Ein Überblick über die derzeit in Europa zugelassenen Basalinsuline gibt Tabelle 1.
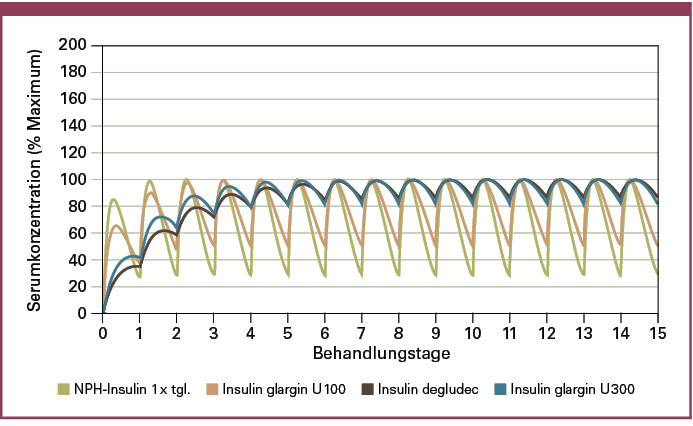
Wie in Abbildung 4 schematisch illustriert, ist die Absorptionsvariabilität eines Insulins direkt abhängig von seiner Halbwertszeit und der Konstanz des Wirkspiegels nach Erreichen stabiler Plasmaspiegel (Steady State). Je länger die Halbwertszeit eines Insulins ist, desto länger dauert es, bis der Steady State erreicht wird. Danach ist der Insulinspiegel bei Insulinen mit langer Halbwertszeit jedoch sehr stabil, und die Variabilität der Insulinwirkung wird minimiert. Länger wirkende Basalinsuline versprechen somit eine verbesserte Titration bei einer Reduktion des Hypoglykämierisikos. Selbst zeitliche Verschiebungen oder der Ausfall einer einzelnen Injektion haben einen deutlich geringeren klinischen Effekt. Die Schwingung der Basalinsulinkurve wird somit durch eine Verlängerung der Halbwertszeit zunehmend gedämpft.
NPH-Insuline
Durch die Bindung an das Eiweiß Protamin (NPH: Neutrales Protamin Hagedorn) wird bei NPH-Insulinen (Protaphane®, Insuman Basal®, Huminsulin Basal®, Berlinsulin H Basal®) eine verzögerte Absorption aus dem subkutanen Fettgewebe erreicht. Durch diesen Verzögerungsmechanismus erreichen NPH-Insuline eine Halbwertszeit von ca. 5 bis 10 Stunden. Aufgrund der relativ kurzen Halbwertszeit ist eine 24-stündige Basalinsulinabdeckung nur mit 2 Injektionen pro Tag möglich. NPH-Insuline erreichen nach 5 bis 7 Stunden ein Wirkmaximum, welches insbesondere bei abendlicher Injektion des NPH-Insulins mit einem gesteigerten Risiko für nächtliche Hypoglykämien einhergeht.
Ein weiteres Problem der kurzen Halbwertszeit ist in einem zu frühen Abklingen der Wirkung mit dem Auftreten von Basalinsulinlücken zu sehen (Abbildung 3). NPH-Insuline unterliegen einer erheblichen Absorptionsvariabilität, die selbst bei ordnungsgemäßer Anwendung beim einzelnen Patienten über 60 % betragen kann (6). NPH-Insuline liegen in einer Suspension vor. Vor Injektion des Insulins muss die Ampulle bzw. der Pen mindestens 20-mal hin- und hergeschwenkt werden, um eine gleichmäßige Suspension des Insulins mit dem NPH zu gewährleisten. Geschieht dies nicht, muss mit einer erheblich höheren Absorptionsvariabilität gerechnet werden. Eine Einschätzung der zu erwartenden Insulinwirkung wird in diesem Fall praktisch unmöglich.
Insulin glargin
Als erstes im Markt verfügbares langwirkendes Insulinanalogon hat Insulin glargin (Lantus®, Abasaglar®) über viele Jahre den Standard der Basalinsulintherapie für Patienten mit Typ-1- und Typ-2-Diabetes geprägt. Dieses Insulinanalogon ist charakterisiert durch zwei zusätzliche Arginine am C-terminalen Ende der B-Kette sowie einem Austausch von Asparagin durch Glycin in Position 21 der A-Kette. Insulin glargin liegt in einer sauren Lösung zur Injektion vor. Nach Injektion des Insulins ins pH-neutrale subkutane Gewebe kommt es zur Ausbildung amorpher Präzipitate. Aus diesen Präzipitaten werden die einzelnen Insulinmoleküle im Vergleich zu NPH-Formulierungen deutlich langsamer in die Blutbahn aufgenommen. Bereits im subkutanen Gewebe und in der Blutzirkulation wird Insulin glargin schnell in die stoffwechselaktiven Metaboliten M1 und M2 gespalten, wobei M1 den für die blutzuckersenkende Wirkung maßgeblichen Metaboliten darstellt (Abbildung 5).
Mit Insulin glargin wurde im Vergleich zu NPH-Insulinen erstmals ein Basalinsulin mit einer 24-stündigen Wirkung und einem deutlich geringeren Wirkmaximum in die Therapie eingeführt. Neben einer Reduktion der Injektionsfrequenz konnte hierdurch eine bessere Einstellung der Nüchternblutzuckerwerte und eine Reduktion des nächtlichen Hypoglykämierisikos im Vergleich zu NPH-Insulinen erreicht werden. Einen wesentlichen Einfluss dürfte hierbei auch eine Reduktion der intraindividuellen Absorptionsvariabilität von Insulin glargin im Vergleich zu NPH-Insulinen haben. Als bisher einziges Insulin kann Insulin glargin eine Endpunktstudie aufweisen (Outcome Reduction with Initial Glargine Intervention [ORIGIN]-Studie), in der die kardiovaskuläre Sicherheit einer Therapie mit diesem Insulin belegt wurde (7). Mit Abasaglar® ist Insulin glargin U 100 als erstes Biosimilar-Insulin in Deutschland verfügbar.
Insulin glargin U 300
Eine weitere Möglichkeit, die Absorptionskinetik eines Insulins aus dem subkutanen Gewebe zu beeinflussen, liegt in einer Veränderung der Konzentration des Insulins in seiner Formulierung. So führt die dreifach höhere Konzentration des Insulin glargin U 300 (Toujeo®) im Vergleich zu Insulin glargin U 100 (Lantus®, Abasaglar®) zu einer Verringerung des Injektionsvolumens. Nach Injektion dieses höher konzentrierten Insulins in das subkutane Fettgewebe bildet sich ein subkutanes Präzipitat, welches eine im Vergleich zu Insulin glargin U 100 um ca. 50 % verkleinerte Oberfläche aufweist und damit eine langsamere Absorption aus dem subkutanen Gewebe gewährleistet.
Wie in Tabelle 1 dargestellt, werden durch diese pharmakologische Modifikation der Insulinformulierung eine längere Halbwertszeit sowie eine weitere Reduktion der Absorptionsvariabilität ohne weitere Modifikation des Insulinmoleküls erreicht. Im klinischen Studienprogramm konnte eine Reduktion nächtlicher Hypoglykämien und eine geringere Gewichtszunahme unter Insulin glargin U 300 im Vergleich zu Insulin glargin U 100 bei Patienten mit Typ-1- und Typ-2-Diabetes gezeigt werden (8). Darüber hinaus erlaubt diese Insulinformulierung eine weitere Flexibilisierung des Insulinzeitpunkts. So zeigte sich eine Variation des Injektionszeitpunkts um ± 3 Stunden unter Insulin glargin U 300 ohne Einfluss auf die Stoffwechselkontrolle. Der in den Zulassungsstudien geringfügig höhere Insulinbedarf (10 – 15 %) unter Insulin glargin U 300 verglichen mit Insulin glargin U 100 wird durch die längere Verweildauer im subkutanen Gewebe und einem dadurch erhöhten lokalen Abbau des Insulins erklärt.
Insulin detemir
Durch Austausch der Aminosäure Threonin in Position B30 und der Ankopplung einer langkettigen Fettsäure in Position B29 wird bei Insulin detemir (Levemir®) eine vermehrte Bindung an das Eiweiß Albumin im subkutanen, intravaskulären und extrazellulären Raum bedingt. Hierdurch kommt es neben einer Veränderung der Absorptionskinetik zu einer zeitlich verzögerten Freigabe des Insulins aus der Albuminbindung im intravasalen und extrazellulären Raum und damit zu einer weiteren Protrahierung der Insulinwirkung. Aufgrund der intermediären Wirkdauer dieses Verzögerungsinsulins ist eine ein- bis zweimalige tägliche Injektion erforderlich. Ein gewisser Nachteil dieses Insulinmoleküls ist in der Reduktion der Affinität zum Insulinrezeptor zu sehen. Hieraus ergibt sich eine verminderte Biopotenz, was wiederum eine ca. 5-fach höhere absolute Insulinmenge im Vergleich zu NPH-Insulin oder Insulin glargin erforderlich macht (1). Klinisch konnte unter Insulin detemir eine im Vergleich zu NPH-Insulin vergleichbare Wirkung bei geringerer Gewichtszunahme demonstriert werden (9).
Insulin degludec
Ähnlich wie Insulin detemir basiert Insulin degludec (Tresiba®) auf einem gering veränderten Humaninsulinmolekül, dem eine langkettige Fettsäure in Position B29 angehängt wurde. Nach Injektion in das Unterhautfettgewebe bilden sich lange Multihexamerketten, aus denen die Insulinmonomere langsam freigesetzt und in die Blutbahn aufgenommen werden. Wie Insulin detemir bindet auch Insulin degludec an Albumin, welches eine protrahierte Abgabe an die peripheren Gewebe (Muskel-, Fettzellen) bedingt und zu einer Reduktion der Wirkvariabilität beiträgt. Nach den vorliegenden pharmakologischen/pharmakodynamischen Untersuchungen weist Insulin degludec unter den in Europa derzeit zugelassenen Insulinen die längste Halbwertszeit und die geringste Absorptionsvariabilität auf (10).
In klinischen Studien mit Typ-1- und Typ-2-Diabetikern konnten mit diesem sehr lang wirkenden Insulinanalogon eine Reduktion des Nüchternblutzuckerwerts und/oder eine Verminderung des nächtlichen Hypoglykämierisikos im Vergleich zu Insulin glargin U 100 dargestellt werden (11, 12). In weiteren Studien wurde der Einfluss variabler Injektionszeiten unter einer Therapie mit Insulin degludec untersucht. Ein Variieren der Injektionszeit in einem Bereich zwischen 8 und 40 Stunden zeigte hierbei keinen Einfluss auf die Qualität der Blutzuckerkontrolle (13, 14). Aufgrund der methodischen Bewertungskriterien des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) und einem zweifelhaften Bewertungsverfahren im AMNOG (Arzneimittelmarktneuordnungsgesetz)-Prozess ist dieses moderne Verzögerungsinsulin allerdings in Deutschland nicht mehr erstattungsfähig.
Mahlzeiteninsuline
Neben einer optimierten Basalinsulinabdeckung ist für Patienten mit Typ-1-Diabetes und für einen Teil der Patienten mit Typ-2-Diabetes die Zufuhr eines Insulins zur Mahlzeit als prandiales Insulin erforderlich. Die Kinetik der subkutanen Substitution von Humaninsulin mit einem Wirkmaximum nach ca. 3 Stunden und einer Wirkdauer von bis zu 8 Stunden hat sich in vielen Fällen als zu langsam erwiesen. Der Nachteil ist in einem unphysiologischen Insulinprofil mit einem zu langsamen und zu lange anhaltenden Anfluten des Insulins aus dem subkutanen Gewebe in die Blutbahn zu sehen. Als Folge des langsamen Wirkeintritts steigen die Blutzuckerwerte nach einer Mahlzeit zu stark an, während die lange Halbwertszeit des Humaninsulins eine erhöhte Hypoglykämiegefahr nach sich zieht. Daraufhin wurden Insulinanaloga entwickelt, die durch eine schnellere Absorption aus dem subkutanen Gewebe eine optimierte prandiale Insulinabdeckung mit einer schneller eintretenden und verkürzten Wirkdauer gewährleisten sollen.
Insulinmoleküle liegen in hoher Konzentration (in der Ampulle oder dem Pen) in Form von Hexameren vor. Hierbei gruppieren sich 6 Insulinmoleküle um 2 Zinkatome. Nach subkutaner Applikation zerfallen diese Komplexe langsam zu Monomeren und freien Zinkatomen. Da lediglich Insulinmonomere die Gefäßwand penetrieren können, ist die Kinetik der Dissoziation der Insulinhexamere im subkutanen Gewebe der zeitlimitierende Schritt der Absorption aus dem subkutanen Gewebe. Die langsame Dissoziationsrate ist daher für die verzögerte und protrahierte Wirkung des Humaninsulins nach subkutaner Injektion verantwortlich. Durch eine Modifikation der Aminosäuresequenz bei den schnellwirkenden Insulinanaloga wurde die Neigung zur Selbstassoziation der Insulinmoleküle herabgesetzt und damit eine schnellere Absorption der Insuline aus dem subkutanen Gewebe erreicht. Somit wird durch die Verwendung schnellwirkender Insulinanaloga eine bessere Annäherung an eine physiologische prandiale Insulinkinetik erreicht (Abbildung 6).
Derzeit stehen 3 schnellwirkende Insulinanaloga (Insulin lispro [Humalog®, Liprolog®], Insulin aspart [NovoRapid®], Insulin glulisin [Apidra®]) für den klinischen Einsatz zur Verfügung. Hierbei wurden bei den einzelnen schnellwirkenden Insulinanaloga folgende Modifikationen der Aminosäuresequenz vorgenommen:
- Insulin lispro: Austausch von Prolin durch Lysin in Position B29,
- Insulin glulisin: Austausch von Asparagin durch Lysin in Position B3 und Austausch von Lysin durch Glutamin in Position B29,
- Insulin aspart: Austausch von Prolin durch Asparagin an Position B28.
Da alle 3 Insulinanaloga eine beschleunigte subkutane Dissoziation der Insulinmoleküle aufweisen, wird die maximale Wirkung bei allen Insulinanaloga schneller als unter einem Humaninsulin erreicht. So wird die maximale Wirkung der Insulinanaloga je nach Untersuchung bereits nach 40 bis 70 Minuten erreicht, während die Wirkdauer mit ca. 4 Stunden kürzer als die der Humaninsuline ist. Die pharmakokinetischen und pharmakodynamischen Unterschiede zwischen den verschiedenen schnellwirkenden Insulinanaloga sind marginal und lediglich für das Insulinanalogon Insulin glulisin wurde ein geringfügig schnellerer Wirkeintritt im Vergleich zu Insulin lispro in Clampuntersuchungen dargestellt (15). Im Gegensatz zu den anderen schnellwirkenden Insulinanaloga ist die Formulierung von Insulin glulisin zinkfrei und enthält Polysorbat 20 zur Stabilisation. Dies könnte möglicherweise eine zusätzliche Verminderung der Selbstassoziation der Insulinmoleküle bei Insulin glulisin bedingen.
Für Patienten mit hohem Insulinbedarf steht jetzt auch eine Insulin-lispro-Formulierung mit höherer Konzentration zur Verfügung: Insulin lispro U 200 (Humalog U 200®, Liprolog U 200®). Durch eine Verdopplung der Insulinkonzentration in dieser Formulierung werden die Injektionsvolumina für die Patienten halbiert.
Neue Insulinentwicklungen
Trotz vieler Neuerungen in der Insulintherapie in den vergangenen Jahrzehnten durch die Etablierung von Insulinanaloga mit veränderten pharmakokinetischen und pharmakodynamischen Eigenschaften, verbesserten Injektionstechniken und zahlreicher neuer Möglichkeiten der Stoffwechselkontrolle bleibt unsere derzeitige Insulinbehandlung weit hinter dem Anspruch einer physiologischen Insulinsubstitution zurück. Eine physiologische Insulinsubstitution und eine normnahe Blutzuckerregulation ist mit den uns derzeit zur Verfügung stehenden Möglichkeiten der Insulintherapie nur näherungsweise möglich und der Bedarf einer Weiterentwicklung der Insulinformulierungen ist groß.
Neue Basalinsuline sollen eine noch langsamere Wirkkinetik aufweisen und die physiologische Wirkung des Insulins in verschiedenen Organsystemen besser imitieren. Für diesen Zweck sind hepatopräferentielle Insuline in der Entwicklung. Ziel dieser Insuline ist es, einen peripher-hepatischen Gradienten in der Insulinwirkung zu schaffen, um hierdurch eine bessere Suppression der hepatischen Glukosefreisetzung einerseits und ein geringeres Hypoglykämierisiko durch eine Begrenzung der peripheren Glukoseutilisation andererseits zu schaffen. Erste diesbezügliche Entwicklungen scheiterten jedoch aufgrund unerwünschter hepatischer Nebenwirkungen und machen weitergehende Forschungen erforderlich.
Neue Mahlzeiteninsuline hingegen sollten sich durch einen möglichst noch schnelleren Wirkeintritt und eine verbesserte Steuerbarkeit auszeichnen. Auch im Hinblick auf potentielle selbstregulierende Insulinpumpensysteme (Closed Loop, artifizielles Pankreas) wäre eine möglichst schnelle Absorption und Steuerbarkeit des subkutan applizierten Insulins von unschätzbarem Wert. Das Insulin Viaject® ist ein neues, schnellwirkendes Insulin, bei dem durch Zusatz von EDTA (ethylene diamine tetraacetic acid, Ethylendiamintetraessigsäure) eine Bindung der Zinkatome erfolgt, was mit einer verminderten Selbstassoziation der Insulinmoleküle einhergeht.
Der Zusatz von Zitrat führt darüber hinaus zu einer Neutralisation der elektrischen Ladungen der Insulinmoleküle. Durch Zugabe des Absorptionsverstärkers Nicotinamid und L-Arginin als Stabilisator wird die Absorption des schnellwirkenden Insulinanalogons Insulin aspart weiter beschleunigt (16). Durch Veränderung der Proteinfaltung mittels Chaperonen konnte die Absorption von Insulin lispro weiter beschleunigt werden. Verschiedene neue, noch schneller wirkende Insulinformulierungen werden in den kommenden Jahren den Markt erreichen und insbesondere im Hinblick auf Closed-Loop-Pumpensysteme ihre klinische Bedeutung finden.
Glukoseabhängig wirkende Insulinformulierungen (Smart-Insuline)
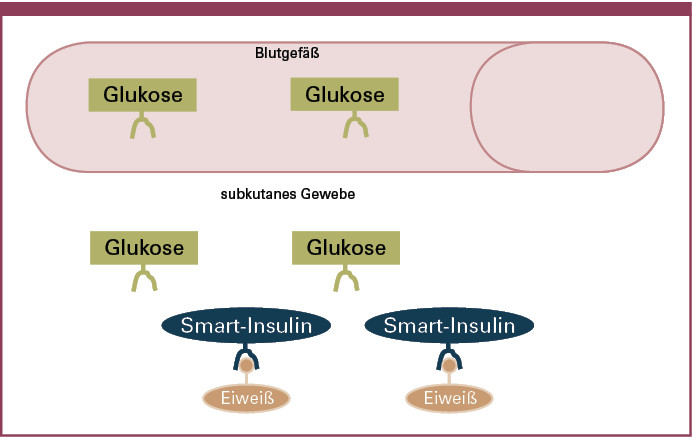
Derzeit werden verschiedene Technologien entwickelt, die eine glukoseabhängige Absorption von Insulin aus dem subkutanen Gewebe erlauben sollen. Bei diesen Insulinen liegt das Insulin an spezielle Trägereiweiße gekoppelt im subkutanen Gewebe vor. Lediglich bei ansteigenden Glukosekonzentrationen werden die Insulinmoleküle kompetitiv von ihren Trägereiweißen verdrängt und diffundieren anschließend in die Blutbahn (Abbildung 7). Mit dieser Technologie würden Insuline geschaffen, die in direkter Abhängigkeit vom Blutzucker ihre Wirkung entfalten. Diese Technologien befinden sich noch in frühen experimentellen Phasen, könnten aber die subkutane Insulinapplikation revolutionieren und zu einem Meilenstein auf dem Weg zur physiologischen Insulinsubstitution werden.
Darüber hinaus werden alternative Applikationswege (oral, buccal, inhalativ u. a.) erforscht. Neben einer veränderten Pharmakokinetik könnte der Vorteil gastrointestinal applizierter Insuline in einer hepatopräferentiellen Wirkung dieser Präparate liegen.
Zusammenfassend werden für die nächsten Jahre zahlreiche Neuentwicklungen in der Insulintherapie erwartet. Eine Annäherung der Kinetik der exogenen Insulinsubstitution an physiologische Sekretions- und Verteilungsmuster im Körper lassen auf eine weitere Optimierung der Blutzuckereinstellung hoffen. Inwieweit potentielle Vorteile neuer Insulintherapien in Deutschland eine angemessene Würdigung erfahren werden, wird der Inhalt zahlreicher Bewertungsverfahren im Rahmen des AMNOG werden. Ob und wann diese neuen Technologien dann auch für deutsche Patienten zur Verfügung stehen werden, hängt nicht nur von der Forschung, sondern weit mehr von gesundheitspolitischen Entscheidungen ab.
Interessenkonflikte: Thomas Forst hat Berater- und Vortragshonorare für wissenschaftliche Veranstaltungen von den Unternehmen Lilly, Sanofi, Berlin-Chemie und Novo Nordisk erhalten.
Johannes Jahnke gibt keinen Interessenkonflikt in Bezug auf die Inhalte des Artikels an.
Dieser CME-Beitrag wurde unterstützt von Sanofi-Aventis Deutschland GmbH.
Auf unserem CME-Portal www.med-etraining.de können Sie ab dem 20.04.2016 diesen Beitrag bearbeiten und bekommen bei Erfolg Ihre Punkte sofort gutgeschrieben.
Erschienen in: Diabetes, Stoffwechsel und Herz, 2016; 26 (2) Seite 108-114